Mississippi Second Chronic Wasting Disease Confirmed in Pontotoc County by National Veterinary Services Laboratory
Mississippi Second Chronic Wasting Disease Confirmed in Pontotoc County by National Veterinary Services Laboratory
Chronic Wasting Disease Confirmed in Pontotoc County
10/30/2018 3:24:47 PM
From MDWFP

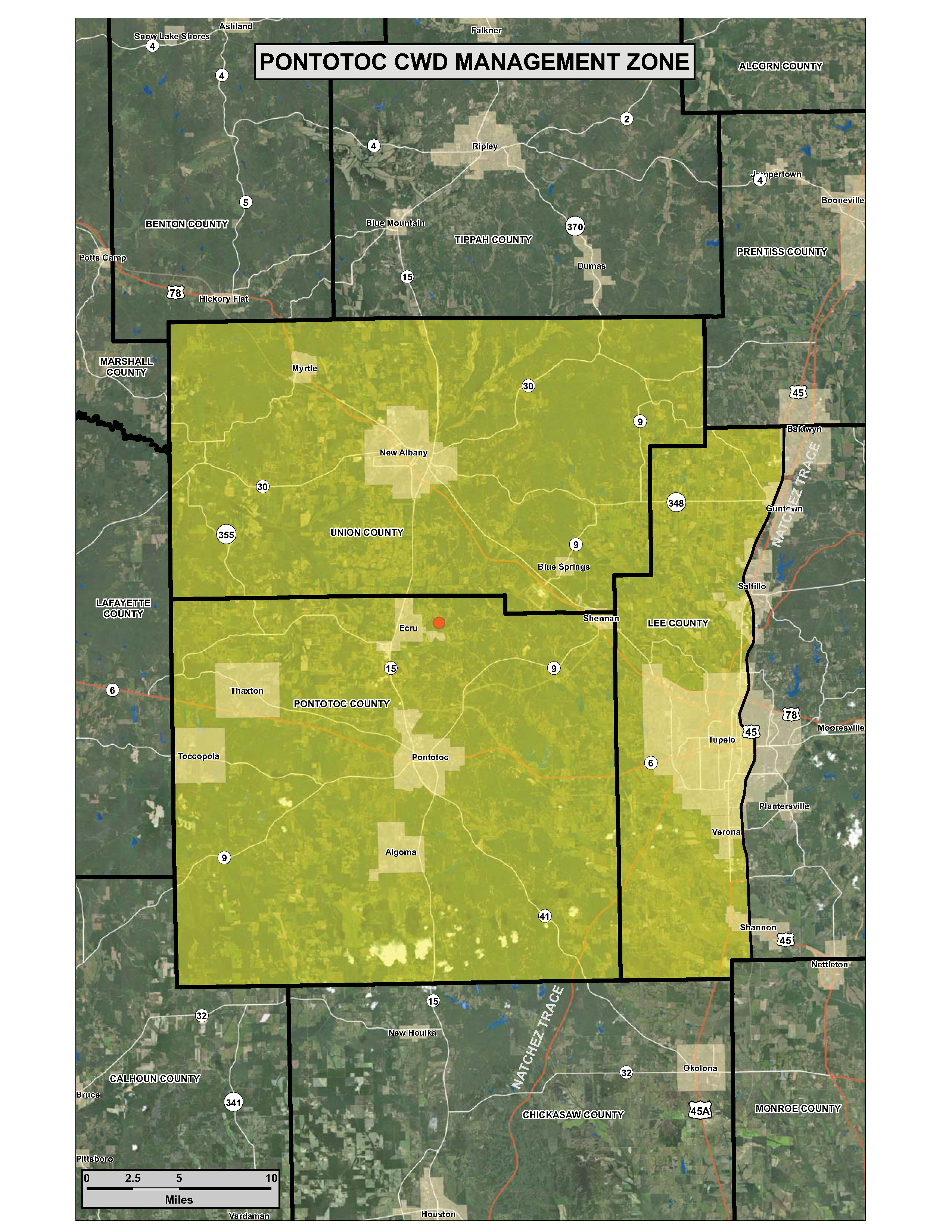
PONTOTOC – The Mississippi Department of Wildlife, Fisheries, and Parks (MDWFP) received confirmation from the National Veterinary Services Laboratory that a white-tailed deer collected in Pontotoc County on October 8, 2018 tested positive for Chronic Wasting Disease (CWD). MDWFP has established the Pontotoc CWD Management Zone that includes Pontotoc and Union counties and all portions of Lee County west of Hwy 45. For any MDWFP-defined CWD Management Zone it is unlawful to:
- Supplemental feed;
- Establish new mineral sites or add supplements to existing sites;
- Remove certain portions of cervid carcasses from the zone (carcass regulations); or
- Trap wild hogs without a permit from MDWFP.
 (click to enlarge)
(click to enlarge)
To monitor CWD in the Pontotoc Zone, MDWFP will rely on hunter-harvested deer during the 2018–19 hunting season. Hunters can submit deer for testing at established drop-off locations or MDWFP-staffed check stations.
MDWFP will host a public meeting to discuss Chronic Wasting Disease (CWD) at North Pontotoc Attendance Center on Thursday, November 8 at 6:00 PM. Presentations by MDWFP staff will be on the status of CWD and planned monitoring activities. Biologists and Law Enforcement officials will be available to answer questions.
For more information about Chronic Wasting Disease visit www.mdwfp.com/cwd. Follow us on Facebook at www.facebook.com/mdwfp or on Twitter at www.twitter.com/MDWFPonline.
Cc: cjdvoice <cjdvoice@yahoogroups.com>; bloodcjd <bloodcjd@yahoogroups.com>
Sent: Fri, Oct 19, 2018 9:00 pm
Subject: Mississippi Chronic Wasting Disease Suspected in a Second White-tailed Deer 10/19/2018 4:03:08 PM From MDWFP
Chronic Wasting Disease Suspected in a Second Mississippi White-tailed Deer 10/19/2018 4:03:08 PM From MDWFP
JACKSON – A 1.5-year-old male white-tailed deer collected on October 8, 2018 in Pontotoc County has tested positive for Chronic Wasting Disease (CWD) from initial testing. This is the second animal in Mississippi testing positive for the disease, which is fatal to white-tailed deer. A sample will be shipped to the National Veterinary Sciences Laboratory in Iowa for an additional, definitive test.
The Mississippi Department of Wildlife, Fisheries, and Parks (MDWFP) will await final results before implementing the agency’s CWD Response Plan. MDWFP encourages hunters to assist with CWD monitoring efforts by voluntarily submitting samples for testing. A list of CWD sample collection locations can be found on www.mdwfp.com/cwd.
For more information regarding CWD in Mississippi, visit our website at www.mdwfp.com/cwd or call us at (601) 432-2199. Follow us on Facebook at www.facebook.com/mdwfp or on Twitter at www.twitter.com/MDWFPonline
FRIDAY, FEBRUARY 09, 2018
Mississippi Chronic Wasting Disease confirmed in a White-tailed Deer
TUESDAY, FEBRUARY 13, 2018
*** MISSISSIPPI STATE DEPARTMENT OF HEALTH Chronic Wasting Disease: Public Health Recommendations ***
***> Terrible news, a study is looking into the fact that CWD has already transmitted to humans as sporadic CJD...see;
Cervid to human prion transmission
Kong, Qingzhong
Case Western Reserve University, Cleveland, OH, United States
Abstract
Prion disease is transmissible and invariably fatal. Chronic wasting disease (CWD) is the prion disease affecting deer, elk and moose, and it is a widespread and expanding epidemic affecting 22 US States and 2 Canadian provinces so far. CWD poses the most serious zoonotic prion transmission risks in North America because of huge venison consumption (>6 million deer/elk hunted and consumed annually in the USA alone), significant prion infectivity in muscles and other tissues/fluids from CWD-affected cervids, and usually high levels of individual exposure to CWD resulting from consumption of the affected animal among often just family and friends. However, we still do not know whether CWD prions can infect humans in the brain or peripheral tissues or whether clinical/asymptomatic CWD zoonosis has already occurred, and we have no essays to reliably detect CWD infection in humans.
We hypothesize that:
(1) The classic CWD prion strain can infect humans at low levels in the brain and peripheral lymphoid tissues;
(2) The cervid-to-human transmission barrier is dependent on the cervid prion strain and influenced by the host (human) prion protein (PrP) primary sequence;
(3) Reliable essays can be established to detect CWD infection in humans; and
(4) CWD transmission to humans has already occurred. We will test these hypotheses in 4 Aims using transgenic (Tg) mouse models and complementary in vitro approaches.
Aim 1 will prove that the classical CWD strain may infect humans in brain or peripheral lymphoid tissues at low levels by conducting systemic bioassays in a set of humanized Tg mouse lines expressing common human PrP variants using a number of CWD isolates at varying doses and routes. Experimental human CWD samples will also be generated for Aim 3.
Aim 2 will test the hypothesis that the cervid-to-human prion transmission barrier is dependent on prion strain and influenced by the host (human) PrP sequence by examining and comparing the transmission efficiency and phenotypes of several atypical/unusual CWD isolates/strains as well as a few prion strains from other species that have adapted to cervid PrP sequence, utilizing the same panel of humanized Tg mouse lines as in Aim 1.
Aim 3 will establish reliable essays for detection and surveillance of CWD infection in humans by examining in details the clinical, pathological, biochemical and in vitro seeding properties of existing and future experimental human CWD samples generated from Aims 1-2 and compare them with those of common sporadic human Creutzfeldt-Jakob disease (sCJD) prions.
Aim 4 will attempt to detect clinical CWD-affected human cases by examining a significant number of brain samples from prion-affected human subjects in the USA and Canada who have consumed venison from CWD-endemic areas utilizing the criteria and essays established in Aim 3. The findings from this proposal will greatly advance our understandings on the potential and characteristics of cervid prion transmission in humans, establish reliable essays for CWD zoonosis and potentially discover the first case(s) of CWD infection in humans.
Public Health Relevance
There are significant and increasing human exposure to cervid prions because chronic wasting disease (CWD, a widespread and highly infectious prion disease among deer and elk in North America) continues spreading and consumption of venison remains popular, but our understanding on cervid-to-human prion transmission is still very limited, raising public health concerns. This proposal aims to define the zoonotic risks of cervid prions and set up and apply essays to detect CWD zoonosis using mouse models and in vitro methods. The findings will greatly expand our knowledge on the potentials and characteristics of cervid prion transmission in humans, establish reliable essays for such infections and may discover the first case(s) of CWD infection in humans.
Funding Agency
Agency
National Institute of Health (NIH)
Institute
National Institute of Neurological Disorders and Stroke (NINDS)
Type
Research Project (R01)
Project #
5R01NS088604-04
Application #
9517118
Study Section
Cellular and Molecular Biology of Neurodegeneration Study Section (CMND)
Program Officer Wong, May
Project Start 2015-09-30 Project End 2019-07-31 Budget Start 2018-08-01 Budget End 2019-07-31 Support Year 4 Fiscal Year 2018 Total Cost Indirect Cost Institution Name Case Western Reserve University Department Pathology Type Schools of Medicine DUNS # 077758407 City Cleveland State OH Country United States Zip Code 44106
Related projects
NIH 2018 R01 NS Cervid to human prion transmission Kong, Qingzhong / Case Western Reserve University
NIH 2017 R01 NS Cervid to human prion transmission Kong, Qingzhong / Case Western Reserve University
NIH 2016 R01 NS Cervid to human prion transmission Kong, Qingzhong / Case Western Reserve University
NIH 2015 R01 NS Cervid to human prion transmission Kong, Qingzhong / Case Western Reserve University $337,507
ZOONOTIC CHRONIC WASTING DISEASE CWD TSE PRION UPDATE
here is the latest;
PRION 2018 CONFERENCE
Oral transmission of CWD into Cynomolgus macaques: signs of atypical disease, prion conversion and infectivity in macaques and bio-assayed transgenic mice
Hermann M. Schatzl, Samia Hannaoui, Yo-Ching Cheng, Sabine Gilch (Calgary Prion Research Unit, University of Calgary, Calgary, Canada) Michael Beekes (RKI Berlin), Walter Schulz-Schaeffer (University of Homburg/Saar, Germany), Christiane Stahl-Hennig (German Primate Center) & Stefanie Czub (CFIA Lethbridge).
To date, BSE is the only example of interspecies transmission of an animal prion disease into humans. The potential zoonotic transmission of CWD is an alarming issue and was addressed by many groups using a variety of in vitro and in vivo experimental systems. Evidence from these studies indicated a substantial, if not absolute, species barrier, aligning with the absence of epidemiological evidence suggesting transmission into humans. Studies in non-human primates were not conclusive so far, with oral transmission into new-world monkeys and no transmission into old-world monkeys.
Our consortium has challenged 18 Cynomolgus macaques with characterized CWD material, focusing on oral transmission with muscle tissue. Some macaques have orally received a total of 5 kg of muscle material over a period of 2 years. After 5-7 years of incubation time some animals showed clinical symptoms indicative of prion disease, and prion neuropathology and PrPSc deposition were detected in spinal cord and brain of some euthanized animals. PrPSc in immunoblot was weakly detected in some spinal cord materials and various tissues tested positive in RT-QuIC, including lymph node and spleen homogenates. To prove prion infectivity in the macaque tissues, we have intracerebrally inoculated 2 lines of transgenic mice, expressing either elk or human PrP. At least 3 TgElk mice, receiving tissues from 2 different macaques, showed clinical signs of a progressive prion disease and brains were positive in immunoblot and RT-QuIC. Tissues (brain, spinal cord and spleen) from these and pre-clinical mice are currently tested using various read-outs and by second passage in mice. Transgenic mice expressing human PrP were so far negative for clear clinical prion disease (some mice >300 days p.i.). In parallel, the same macaque materials are inoculated into bank voles.
Taken together, there is strong evidence of transmissibility of CWD orally into macaques and from macaque tissues into transgenic mouse models, although with an incomplete attack rate. The clinical and pathological presentation in macaques was mostly atypical, with a strong emphasis on spinal cord pathology. Our ongoing studies will show whether the transmission of CWD into macaques and passage in transgenic mice represents a form of non-adaptive prion amplification, and whether macaque-adapted prions have the potential to infect mice expressing human PrP. The notion that CWD can be transmitted orally into both new-world and old-world non-human primates asks for a careful reevaluation of the zoonotic risk of CWD.
***> The notion that CWD can be transmitted orally into both new-world and old-world non-human primates asks for a careful reevaluation of the zoonotic risk of CWD. <***
READING OVER THE PRION 2018 ABSTRACT BOOK, LOOKS LIKE THEY FOUND THAT from this study ;
P190 Human prion disease mortality rates by occurrence of chronic wasting disease in freeranging cervids, United States
Abrams JY (1), Maddox RA (1), Schonberger LB (1), Person MK (1), Appleby BS (2), Belay ED (1) (1) Centers for Disease Control and Prevention (CDC), National Center for Emerging and Zoonotic Infectious Diseases, Atlanta, GA, USA (2) Case Western Reserve University, National Prion Disease Pathology Surveillance Center (NPDPSC), Cleveland, OH, USA.
SEEMS THAT THEY FOUND Highly endemic states had a higher rate of prion disease mortality compared to non-CWD states.
AND ANOTHER STUDY;
P172 Peripheral Neuropathy in Patients with Prion Disease
Wang H(1), Cohen M(1), Appleby BS(1,2) (1) University Hospitals Cleveland Medical Center, Cleveland, Ohio (2) National Prion Disease Pathology Surveillance Center, Cleveland, Ohio..
IN THIS STUDY, THERE WERE autopsy-proven prion cases from the National Prion Disease Pathology Surveillance Center that were diagnosed between September 2016 to March 2017, AND included 104 patients.
SEEMS THEY FOUND THAT The most common sCJD subtype was MV1-2 (30%), followed by MM1-2 (20%), AND THAT The Majority of cases were male (60%), AND half of them had exposure to wild game.
snip...see more on Prion 2017 Macaque study from Prion 2017 Conference and other updated science on cwd tse prion zoonosis below...terry
Prion 2017
Conference Abstracts CWD 2017 PRION CONFERENCE
First evidence of intracranial and peroral transmission of Chronic Wasting Disease (CWD) into Cynomolgus macaques: a work in progress
Stefanie Czub1, Walter Schulz-Schaeffer2, Christiane Stahl-Hennig3, Michael Beekes4, Hermann Schaetzl5 and Dirk Motzkus6 1
University of Calgary Faculty of Veterinary Medicine/Canadian Food Inspection Agency; 2Universitatsklinikum des Saarlandes und Medizinische Fakultat der Universitat des Saarlandes; 3 Deutsches Primaten Zentrum/Goettingen; 4 Robert-Koch-Institut Berlin; 5 University of Calgary Faculty of Veterinary Medicine; 6 presently: Boehringer Ingelheim Veterinary Research Center; previously: Deutsches Primaten Zentrum/Goettingen
This is a progress report of a project which started in 2009. 21 cynomolgus macaques were challenged with characterized CWD material from white-tailed deer (WTD) or elk by intracerebral (ic), oral, and skin exposure routes. Additional blood transfusion experiments are supposed to assess the CWD contamination risk of human blood product. Challenge materials originated from symptomatic cervids for ic, skin scarification and partially per oral routes (WTD brain). Challenge material for feeding of muscle derived from preclinical WTD and from preclinical macaques for blood transfusion experiments. We have confirmed that the CWD challenge material contained at least two different CWD agents (brain material) as well as CWD prions in muscle-associated nerves. Here we present first data on a group of animals either challenged ic with steel wires or per orally and sacrificed with incubation times ranging from 4.5 to 6.9 years at postmortem. Three animals displayed signs of mild clinical disease, including anxiety, apathy, ataxia and/or tremor. In four animals wasting was observed, two of those had confirmed diabetes. All animals have variable signs of prion neuropathology in spinal cords and brains and by supersensitive IHC, reaction was detected in spinal cord segments of all animals. Protein misfolding cyclic amplification (PMCA), real-time quaking-induced conversion (RT-QuiC) and PET-blot assays to further substantiate these findings are on the way, as well as bioassays in bank voles and transgenic mice. At present, a total of 10 animals are sacrificed and read-outs are ongoing. Preclinical incubation of the remaining macaques covers a range from 6.4 to 7.10 years. Based on the species barrier and an incubation time of > 5 years for BSE in macaques and about 10 years for scrapie in macaques, we expected an onset of clinical disease beyond 6 years post inoculation.
PRION 2017
DECIPHERING NEURODEGENERATIVE DISORDERS
Subject: PRION 2017 CONFERENCE DECIPHERING NEURODEGENERATIVE DISORDERS VIDEO
PRION 2017
CONFERENCE DECIPHERING NEURODEGENERATIVE DISORDERS
*** PRION 2017 CONFERENCE VIDEO
TUESDAY, JUNE 13, 2017
PRION 2017 CONFERENCE ABSTRACT
First evidence of intracranial and peroral transmission of Chronic Wasting Disease (CWD) into Cynomolgus macaques: a work in progress
SATURDAY, JULY 29, 2017
Risk Advisory Opinion: Potential Human Health Risks from Chronic Wasting Disease CFIA, PHAC, HC (HPFB and FNIHB), INAC, Parks Canada, ECCC and AAFC
just out CDC...see;
Volume 24, Number 8—August 2018
Research
Susceptibility of Human Prion Protein to Conversion by Chronic Wasting Disease Prions
Marcelo A. BarriaComments to Author , Adriana Libori, Gordon Mitchell, and Mark W. Head Author affiliations: National CJD Research and Surveillance Unit, University of Edinburgh, Edinburgh, Scotland, UK (M.A. Barria, A. Libori, M.W. Head); National and OIE Reference Laboratory for Scrapie and CWD, Canadian Food Inspection Agency, Ottawa, Ontario, Canada (G. Mitchell)
M. A. Barria et al.
ABSTRACT
Chronic wasting disease (CWD) is a contagious and fatal neurodegenerative disease and a serious animal health issue for deer and elk in North America. The identification of the first cases of CWD among free-ranging reindeer and moose in Europe brings back into focus the unresolved issue of whether CWD can be zoonotic like bovine spongiform encephalopathy. We used a cell-free seeded protein misfolding assay to determine whether CWD prions from elk, white-tailed deer, and reindeer in North America can convert the human prion protein to the disease-associated form. We found that prions can convert, but the efficiency of conversion is affected by polymorphic variation in the cervid and human prion protein genes. In view of the similarity of reindeer, elk, and white-tailed deer in North America to reindeer, red deer, and roe deer, respectively, in Europe, a more comprehensive and thorough assessment of the zoonotic potential of CWD might be warranted.
snip...
Discussion
Characterization of the transmission properties of CWD and evaluation of their zoonotic potential are important for public health purposes. Given that CWD affects several members of the family Cervidae, it seems reasonable to consider whether the zoonotic potential of CWD prions could be affected by factors such as CWD strain, cervid species, geographic location, and Prnp–PRNP polymorphic variation. We have previously used an in vitro conversion assay (PMCA) to investigate the susceptibility of the human PrP to conversion to its disease-associated form by several animal prion diseases, including CWD (15,16,22). The sensitivity of our molecular model for the detection of zoonotic conversion depends on the combination of 1) the action of proteinase K to degrade the abundant human PrPC that constitutes the substrate while only N terminally truncating any human PrPres produced and 2) the presence of the 3F4 epitope on human but not cervid PrP. In effect, this degree of sensitivity means that any human PrPres formed during the PMCA reaction can be detected down to the limit of Western blot sensitivity. In contrast, if other antibodies that detect both cervid and human PrP are used, such as 6H4, then newly formed human PrPres must be detected as a measurable increase in PrPres over the amount remaining in the reaction product from the cervid seed. Although best known for the efficient amplification of prions in research and diagnostic contexts, the variation of the PMCA method employed in our study is optimized for the definitive detection of zoonotic reaction products of inherently inefficient conversion reactions conducted across species barriers. By using this system, we previously made and reported the novel observation that elk CWD prions could convert human PrPC from human brain and could also convert recombinant human PrPC expressed in transgenic mice and eukaryotic cell cultures (15).
A previous publication suggested that mule deer PrPSc was unable to convert humanized transgenic substrate in PMCA assays (23) and required a further step of in vitro conditioning in deer substrate PMCA before it was able to cross the deer–human molecular barrier (24). However, prions from other species, such as elk (15) and reindeer affected by CWD, appear to be compatible with the human protein in a single round of amplification (as shown in our study). These observations suggest that different deer species affected by CWD could present differing degrees of the olecular compatibility with the normal form of human PrP.
The contribution of the polymorphism at codon 129 of the human PrP gene has been extensively studied and is recognized as a risk factor for Creutzfeldt-Jakob disease (4). In cervids, the equivalent codon corresponds to the position 132 encoding methionine or leucine. This polymorphism in the elk gene has been shown to play an important role in CWD susceptibility (25,26). We have investigated the effect of this cervid Prnp polymorphism on the conversion of the humanized transgenic substrate according to the variation in the equivalent PRNP codon 129 polymorphism. Interestingly, only the homologs methionine homozygous seed–substrate reactions could readily convert the human PrP, whereas the heterozygous elk PrPSc was unable to do so, even though comparable amounts of PrPres were used to seed the reaction. In addition, we observed only low levels of human PrPres formation in the reactions seeded with the homozygous methionine (132 MM) and the heterozygous (132 ML) seeds incubated with the other 2 human polymorphic substrates (129 MV and 129 VV). The presence of the amino acid leucine at position 132 of the elk Prnp gene has been attributed to a lower degree of prion conversion compared with methionine on the basis of experiments in mice made transgenic for these polymorphic variants (26). Considering the differences observed for the amplification of the homozygous human methionine substrate by the 2 polymorphic elk seeds (MM and ML), reappraisal of the susceptibility of human PrPC by the full range of cervid polymorphic variants affected by CWD would be warranted.
In light of the recent identification of the first cases of CWD in Europe in a free-ranging reindeer (R. tarandus) in Norway (2), we also decided to evaluate the in vitro conversion potential of CWD in 2 experimentally infected reindeer (18). Formation of human PrPres was readily detectable after a single round of PMCA, and in all 3 humanized polymorphic substrates (MM, MV, and VV). This finding suggests that CWD prions from reindeer could be more compatible with human PrPC generally and might therefore present a greater risk for zoonosis than, for example, CWD prions from white-tailed deer. A more comprehensive comparison of CWD in the affected species, coupled with the polymorphic variations in the human and deer PRNP–Prnp genes, in vivo and in vitro, will be required before firm conclusions can be drawn. Analysis of the Prnp sequence of the CWD reindeer in Norway was reported to be identical to the specimens used in our study (2). This finding raises the possibility of a direct comparison of zoonotic potential between CWD acquired in the wild and that produced in a controlled laboratory setting. (Table).
The prion hypothesis proposes that direct molecular interaction between PrPSc and PrPC is necessary for conversion and prion replication. Accordingly, polymorphic variants of the PrP of host and agent might play a role in determining compatibility and potential zoonotic risk. In this study, we have examined the capacity of the human PrPC to support in vitro conversion by elk, white-tailed deer, and reindeer CWD PrPSc. Our data confirm that elk CWD prions can convert the human PrPC, at least in vitro, and show that the homologous PRNP polymorphisms at codon 129 and 132 in humans and cervids affect conversion efficiency. Other species affected by CWD, particularly caribou or reindeer, also seem able to convert the human PrP. It will be important to determine whether other polymorphic variants found in other CWD-affected Cervidae or perhaps other factors (17) exert similar effects on the ability to convert human PrP and thus affect their zoonotic potential.
Dr. Barria is a research scientist working at the National CJD Research and Surveillance Unit, University of Edinburgh. His research has focused on understanding the molecular basis of a group of fatal neurologic disorders called prion diseases.
Acknowledgments
snip...see;
Molecular Barriers to Zoonotic Transmission of Prions
Marcelo A. Barria, Aru Balachandran, Masanori Morita, Tetsuyuki Kitamoto, Rona Barron, Jean Manson, Richard Knight, James W. Ironside, and Mark W. Headcorresponding author
snip...
The conversion of human PrPC by CWD brain homogenate in PMCA reactions was less efficient when the amino acid at position 129 was valine rather than methionine.
***Furthermore, the form of human PrPres produced in this in vitro assay when seeded with CWD, resembles that found in the most common human prion disease, namely sCJD of the MM1 subtype.
snip...
However, we can say with confidence that under the conditions used here, none of the animal isolates tested were as efficient as C-type BSE in converting human PrPC, which is reassuring.
***Less reassuring is the finding that there is no absolute barrier to the conversion of human PrPC by CWD prions in a protocol using a single round of PMCA and an entirely human substrate prepared from the target organ of prion diseases, the brain.
ZOONOTIC, ZOONOSIS, CHRONIC WASTING DISEASE CWD TRANSMISSIBLE SPONGIFORM ENCEPHALOPATHY TSE PRION
10. ZOONOTIC, ZOONOSIS, CHRONIC WASTING DISEASE CWD TRANSMISSIBLE SPONGIFORM ENCEPHALOPATHY TSE PRION AKA MAD DEER ELK DISEASE IN HUMANS, has it already happened, that should be the question...
''In particular the US data do not clearly exclude the possibility of human (sporadic or familial) TSE development due to consumption of venison. The Working Group thus recognizes a potential risk to consumers if a TSE would be present in European cervids.'' Scientific opinion on chronic wasting disease (II)
EFSA Panel on Biological Hazards (BIOHAZ) Antonia Ricci Ana Allende Declan Bolton Marianne Chemaly Robert Davies Pablo Salvador Fernández Escámez ... See all authors
First published: 17 January 2018 https://doi.org/10.2903/j.efsa.2018.5132 ;
also, see;
8. Even though human TSE‐exposure risk through consumption of game from European cervids can be assumed to be minor, if at all existing, no final conclusion can be drawn due to the overall lack of scientific data. In particular the US data do not clearly exclude the possibility of human (sporadic or familial) TSE development due to consumption of venison. The Working Group thus recognizes a potential risk to consumers if a TSE would be present in European cervids. It might be prudent considering appropriate measures to reduce such a risk, e.g. excluding tissues such as CNS and lymphoid tissues from the human food chain, which would greatly reduce any potential risk for consumers. However, it is stressed that currently, no data regarding a risk of TSE infections from cervid products are available.
snip...
The tissue distribution of infectivity in CWD‐infected cervids is now known to extend beyond CNS and lymphoid tissues. While the removal of these specific tissues from the food chain would reduce human dietary exposure to infectivity, exclusion from the food chain of the whole carcass of any infected animal would be required to eliminate human dietary exposure.
https://efsa.onlinelibrary..wiley.com/doi/full/10.2903/j.efsa.2018.5132
zoonosis zoonotic cervid tse prion cwd to humans, preparing for the storm
***An alternative to modeling the species barrier is the cell-free conversion assay which points to CWD as the animal prion disease with the greatest zoonotic potential, after (and very much less than) BSE.116***
https://www.tandfonline.com/doi/pdf/10.4161/pri.29237
To date there is no direct evidence that CWD has been or can be transmitted from animals to humans.
However, initial findings from a laboratory research project funded by the Alberta Prion Research Institute (APRI) and Alberta Livestock Meat Agency (ALMA), and led by a Canadian Food Inspection Agency (CFIA) scientist indicate that CWD has been transmitted to cynomolgus macaques (the non-human primate species most closely related to humans that may be used in research), through both the intracranial and oral routes of exposure.
Both infected brain and muscle tissues were found to transmit disease.
Health Canada’s Health Products and Food Branch (HPFB) was asked to consider the impact of these findings on the Branch’s current position on CWD in health products and foods.
Summary and Recommendation:
snip...
Health Portfolio partners were recently made aware of initial findings from a research project led by a CFIA scientist that have demonstrated that cynomolgus macaques can be infected via intracranial exposure and oral gavage with CWD infected muscle.
These findings suggest that CWD, under specific experimental conditions, has the potential to cross the human species barrier, including by enteral feeding of CWD infected muscle.
https://www.thetyee.ca/Documents/2017/06/24/Risk-Advisory-Opinion-CWD-2017.pdf
*** WDA 2016 NEW YORK ***
We found that CWD adapts to a new host more readily than BSE and that human PrP was unexpectedly prone to misfolding by CWD prions.
In addition, we investigated the role of specific regions of the bovine, deer and human PrP protein in resistance to conversion by prions from another species.
***We have concluded that the human protein has a region that confers unusual susceptibility to conversion by CWD prions.
Student Presentations Session 2
The species barriers and public health threat of CWD and BSE prions
Ms. Kristen Davenport1, Dr. Davin Henderson1, Dr. Candace Mathiason1, Dr. Edward Hoover1 1Colorado State University
Chronic wasting disease (CWD) is spreading rapidly through cervid populations in the USA. Bovine spongiform encephalopathy (BSE, mad cow disease) arose in the 1980s because cattle were fed recycled animal protein.
These and other prion diseases are caused by abnormal folding of the normal prion protein (PrP) into a disease causing form (PrPd), which is pathogenic to nervous system cells and can cause subsequent PrP to misfold. CWD spreads among cervids very efficiently, but it has not yet infected humans. On the other hand, BSE was spread only when cattle consumed infected bovine or ovine tissue, but did infect humans and other species.
The objective of this research is to understand the role of PrP structure in cross-species infection by CWD and BSE. To study the propensity of each species’ PrP to be induced to misfold by the presence of PrPd from verious species, we have used an in vitro system that permits detection of PrPd in real-time.
We measured the conversion efficiency of various combinations of PrPd seeds and PrP substrate combinations.
We observed the cross-species behavior of CWD and BSE, in addition to feline-adapted CWD and BSE. We found that CWD adapts to a new host more readily than BSE and that human PrP was unexpectedly prone to misfolding by CWD prions. In addition, we investigated the role of specific regions of the bovine, deer and human PrP protein in resistance to conversion by prions from another species.
***We have concluded that the human protein has a region that confers unusual susceptibility to conversion by CWD prions. CWD is unique among prion diseases in its rapid spread in natural populations. BSE prions are essentially unaltered upon passage to a new species, while CWD adapts to the new species. This adaptation has consequences for surveillance of humans exposed to CWD. Wildlife Disease Risk Communication Research Contributes to Wildlife Trust Administration Exploring perceptions about chronic wasting disease risks among wildlife and agriculture professionals and stakeholders
http://www.wda2016.org/uploads/5/8/6/1/58613359/wda_2016_conference_proceedings_low_res.pdf
We found that CWD adapts to a new host more readily than BSE and that human PrP was unexpectedly prone to misfolding by CWD prions.
In addition, we investigated the role of specific regions of the bovine, deer and human PrP protein in resistance to conversion by prions from another species.
***We have concluded that the human protein has a region that confers unusual susceptibility to conversion by CWD prions.
Student Presentations Session 2
The species barriers and public health threat of CWD and BSE prions
Ms. Kristen Davenport1, Dr. Davin Henderson1, Dr. Candace Mathiason1, Dr. Edward Hoover1 1Colorado State University
Chronic wasting disease (CWD) is spreading rapidly through cervid populations in the USA. Bovine spongiform encephalopathy (BSE, mad cow disease) arose in the 1980s because cattle were fed recycled animal protein.
These and other prion diseases are caused by abnormal folding of the normal prion protein (PrP) into a disease causing form (PrPd), which is pathogenic to nervous system cells and can cause subsequent PrP to misfold. CWD spreads among cervids very efficiently, but it has not yet infected humans. On the other hand, BSE was spread only when cattle consumed infected bovine or ovine tissue, but did infect humans and other species.
The objective of this research is to understand the role of PrP structure in cross-species infection by CWD and BSE. To study the propensity of each species’ PrP to be induced to misfold by the presence of PrPd from verious species, we have used an in vitro system that permits detection of PrPd in real-time.
We measured the conversion efficiency of various combinations of PrPd seeds and PrP substrate combinations.
We observed the cross-species behavior of CWD and BSE, in addition to feline-adapted CWD and BSE. We found that CWD adapts to a new host more readily than BSE and that human PrP was unexpectedly prone to misfolding by CWD prions. In addition, we investigated the role of specific regions of the bovine, deer and human PrP protein in resistance to conversion by prions from another species.
***We have concluded that the human protein has a region that confers unusual susceptibility to conversion by CWD prions. CWD is unique among prion diseases in its rapid spread in natural populations. BSE prions are essentially unaltered upon passage to a new species, while CWD adapts to the new species. This adaptation has consequences for surveillance of humans exposed to CWD. Wildlife Disease Risk Communication Research Contributes to Wildlife Trust Administration Exploring perceptions about chronic wasting disease risks among wildlife and agriculture professionals and stakeholders
http://www.wda2016.org/uploads/5/8/6/1/58613359/wda_2016_conference_proceedings_low_res.pdf
CDC CWD 2018 TRANSMISSION
https://www.cdc.gov/prions/cwd/transmission.html
https://www.cdc.gov/prions/cwd/transmission.html
TUESDAY, SEPTEMBER 12, 2017
CDC Now Recommends Strongly consider having the deer or elk tested for CWD before you eat the meat
http://chronic-wasting-disease.blogspot.com/2017/09/cdc-now-recommends-strongly-consider.html
CDC Now Recommends Strongly consider having the deer or elk tested for CWD before you eat the meat
http://chronic-wasting-disease.blogspot.com/2017/09/cdc-now-recommends-strongly-consider.html
SATURDAY, JANUARY 27, 2018
CDC CHRONIC WASTING DISEASE CWD TSE PRION UPDATE REPORT USA JANUARY 2018
http://chronic-wasting-disease.blogspot.com/2018/01/cdc-chronic-wasting-disease-cwd-tse.html
Subject: CDC CHRONIC WASTING DISEASE CWD TSE PRION UPDATE REPORT USA JANUARY 2018
CHRONIC WASTING DISEASE CWD TSE PRION IS THE USA AND NORTH AMERICA'S MAD COW DISEASE.
THE USDA INC ET AL WORKED VERY HARD CONCEALING BSE TSE PRION IN CATTLE. they almost succeeded $$$
BUT CWD TSE PRION IN CERVIDS IS A DIFFERENT BEAST, THE COVER UP THERE, USDA INC COULD NOT CONTAIN.
SPORADIC CJD IS 85%+ OF ALL HUMAN TSE PRION DISEASE.
SPORADIC CJD HAS NOW BEEN LINKED TO TYPICAL AND ATYPICAL BSE, SCRAPIE, AND CWD.
SPORADIC/SPONTANEOUS TSE HAS NEVER BEEN PROVEN.
***Moreover, sporadic disease has never been observed in breeding colonies or primate research laboratories, most notably among hundreds of animals over several decades of study at the National Institutes of Health25, and in nearly twenty older animals continuously housed in our own facility.***
https://www.nature.com/articles/srep11573
CHRONIC WASTING DISEASE CWD TSE PRION IS THE USA AND NORTH AMERICA'S MAD COW DISEASE.
THE USDA INC ET AL WORKED VERY HARD CONCEALING BSE TSE PRION IN CATTLE. they almost succeeded $$$
BUT CWD TSE PRION IN CERVIDS IS A DIFFERENT BEAST, THE COVER UP THERE, USDA INC COULD NOT CONTAIN.
SPORADIC CJD IS 85%+ OF ALL HUMAN TSE PRION DISEASE.
SPORADIC CJD HAS NOW BEEN LINKED TO TYPICAL AND ATYPICAL BSE, SCRAPIE, AND CWD.
SPORADIC/SPONTANEOUS TSE HAS NEVER BEEN PROVEN.
***Moreover, sporadic disease has never been observed in breeding colonies or primate research laboratories, most notably among hundreds of animals over several decades of study at the National Institutes of Health25, and in nearly twenty older animals continuously housed in our own facility.***
https://www.nature.com/articles/srep11573
CDC CWD TSE PRION UPDATE USA JANUARY 2018
As of January 2018, CWD in free-ranging deer, elk and/or moose has been reported in at least 22 states in the continental United States, as well as two provinces in Canada. In addition, CWD has been reported in reindeer and moose in Norway, and a small number of imported cases have been reported in South Korea. The disease has also been found in farmed deer and elk. CWD was first identified in captive deer in the late 1960s in Colorado and in wild deer in 1981. By the 1990s, it had been reported in surrounding areas in northern Colorado and southern Wyoming. Since 2000, the area known to be affected by CWD in free-ranging animals has increased to at least 22 states, including states in the Midwest, Southwest, and limited areas on the East Coast.. It is possible that CWD may also occur in other states without strong animal surveillance systems, but that cases haven’t been detected yet. Once CWD is established in an area, the risk can remain for a long time in the environment. The affected areas are likely to continue to expand. Nationwide, the overall occurrence of CWD in free-ranging deer and elk is relatively low. However, in several locations where the disease is established, infection rates may exceed 10 percent (1 in 10), and localized infection rates of more than 25 percent (1 in 4) have been reported. The infection rates among some captive deer can be much higher, with a rate of 79% (nearly 4 in 5) reported from at least one captive herd. As of January 2018, there were 186 counties in 22 states with reported CWD in free-ranging cervids...
Chronic Wasting Disease Among Free-Ranging Cervids by County, United States, January 2018
snip....
https://www.cdc.gov/prions/cwd/occurrence.html
*** 2017-2018 CWD TSE Prion UPDATE
https://www.cdc.gov/prions/cwd/occurrence.html
*** The potential impact of prion diseases on human health was greatly magnified by the recognition that interspecies transfer of BSE to humans by beef ingestion resulted in vCJD. While changes in animal feed constituents and slaughter practices appear to have curtailed vCJD, there is concern that CWD of free-ranging deer and elk in the U.S. might also cross the species barrier. Thus, consuming venison could be a source of human prion disease. Whether BSE and CWD represent interspecies scrapie transfer or are newly arisen prion diseases is unknown. Therefore, the possibility of transmission of prion disease through other food animals cannot be ruled out. There is evidence that vCJD can be transmitted through blood transfusion. There is likely a pool of unknown size of asymptomatic individuals infected with vCJD, and there may be asymptomatic individuals infected with the CWD equivalent. These circumstances represent a potential threat to blood, blood products, and plasma supplies.
http://cdmrp.army.mil/prevfunded/nprp/NPRP_Summit_Final_Report.pdf
http://cdmrp.army.mil/prevfunded/nprp/NPRP_Summit_Final_Report.pdf
Transmission Studies
Mule deer transmissions of CWD were by intracerebral inoculation and compared with natural cases {the following was written but with a single line marked through it ''first passage (by this route)}....TSS
resulted in a more rapidly progressive clinical disease with repeated episodes of synocopy ending in coma. One control animal became affected, it is believed through contamination of inoculum (?saline). Further CWD transmissions were carried out by Dick Marsh into ferret, mink and squirrel monkey. Transmission occurred in ALL of these species with the shortest incubation period in the ferret.
snip...
https://web.archive.org/web/20090506002237/http://www.bseinquiry.gov.uk/files/mb/m11b/tab01.pdf
http://www.fsis.usda.gov/OPPDE/Comments/03-025IFA/03-025IFA-2.pdf
Prion Infectivity in Fat of Deer with Chronic Wasting Disease▿
Brent Race#, Kimberly Meade-White#, Richard Race and Bruce Chesebro* + Author Affiliations
In mice, prion infectivity was recently detected in fat. Since ruminant fat is consumed by humans and fed to animals, we determined infectivity titers in fat from two CWD-infected deer. Deer fat devoid of muscle contained low levels of CWD infectivity and might be a risk factor for prion infection of other species.
http://jvi.asm.org/content/83/18/9608.full
Prions in Skeletal Muscles of Deer with Chronic Wasting Disease
Here bioassays in transgenic mice expressing cervid prion protein revealed the presence of infectious prions in skeletal muscles of CWD-infected deer, demonstrating that humans consuming or handling meat from CWD-infected deer are at risk to prion exposure.
http://science.sciencemag.org/content/311/5764/1117.long
*** now, let’s see what the authors said about this casual link, personal communications years ago, and then the latest on the zoonotic potential from CWD to humans from the TOKYO PRION 2016 CONFERENCE.
see where it is stated NO STRONG evidence. so, does this mean there IS casual evidence ???? “Our conclusion stating that we found no strong evidence of CWD transmission to humans”
From: TSS (216-119-163-189.ipset45.wt.net)
Subject: CWD aka MAD DEER/ELK TO HUMANS ???
Date: September 30, 2002 at 7:06 am PST
From: "Belay, Ermias"
To: Cc: "Race, Richard (NIH)" ; ; "Belay, Ermias"
Sent: Monday, September 30, 2002 9:22 AM
Subject: RE: TO CDC AND NIH - PUB MED- 3 MORE DEATHS - CWD - YOUNG HUNTERS
Dear Sir/Madam,
In the Archives of Neurology you quoted (the abstract of which was attached to your email), we did not say CWD in humans will present like variant CJD.. That assumption would be wrong. I encourage you to read the whole article and call me if you have questions or need more clarification (phone: 404-639-3091). Also, we do not claim that "no-one has ever been infected with prion disease from eating venison." Our conclusion stating that we found no strong evidence of CWD transmission to humans in the article you quoted or in any other forum is limited to the patients we investigated.
Ermias Belay, M.D. Centers for Disease Control and Prevention
-----Original Message-----
From: Sent: Sunday, September 29, 2002 10:15 AM
To: rr26k@nih.gov; rrace@niaid.nih.gov; ebb8@CDC.GOV
Subject: TO CDC AND NIH - PUB MED- 3 MORE DEATHS - CWD - YOUNG HUNTERS
Sunday, November 10, 2002 6:26 PM ......snip........end..............TSS
Thursday, April 03, 2008
A prion disease of cervids: Chronic wasting disease 2008 1: Vet Res. 2008 Apr 3;39(4):41 A prion disease of cervids: Chronic wasting disease Sigurdson CJ.
snip...
*** twenty-seven CJD patients who regularly consumed venison were reported to the Surveillance Center***,
snip... full text ;
http://chronic-wasting-disease.blogspot.com/2008/04/prion-disease-of-cervids-chronic.html
> However, to date, no CWD infections have been reported in people.
key word here is 'reported'. science has shown that CWD in humans will look like sporadic CJD. SO, how can one assume that CWD has not already transmitted to humans? they can't, and it's as simple as that. from all recorded science to date, CWD has already transmitted to humans, and it's being misdiagnosed as sporadic CJD. ...terry
*** LOOKING FOR CWD IN HUMANS AS nvCJD or as an ATYPICAL CJD, LOOKING IN ALL THE WRONG PLACES $$$ ***
*** These results would seem to suggest that CWD does indeed have zoonotic potential, at least as judged by the compatibility of CWD prions and their human PrPC target. Furthermore, extrapolation from this simple in vitro assay suggests that if zoonotic CWD occurred, it would most likely effect those of the PRNP codon 129-MM genotype and that the PrPres type would be similar to that found in the most common subtype of sCJD (MM1).***
http://www.tandfonline.com/doi/full/10.4161/pri.28124?src=recsys
http://www.tandfonline.com/doi/pdf/10.4161/pri.28124?needAccess=true
https://wwwnc.cdc.gov/eid/article/20/1/13-0858_article
SEE; Travel History, Hunting, and Venison Consumption Related to Prion Disease Exposure, 2006-2007 FoodNet Population Survey
Monday, May 23, 2011
CDC Assesses Potential Human Exposure to Prion Diseases Travel Warning
Public release date: 23-May-2011
Contact: Francesca Costanzo adajmedia@elsevier.com 215-239-3249 Elsevier Health Sciences
CDC assesses potential human exposure to prion diseases Study results reported in the Journal of the American Dietetic Association Philadelphia, PA, May 23, 2011 – Researchers from the Centers for Disease Control and Prevention (CDC) have examined the potential for human exposure to prion diseases, looking at hunting, venison consumption, and travel to areas in which prion diseases have been reported in animals. Three prion diseases in particular – bovine spongiform encephalopathy (BSE or “Mad Cow Disease”), variant Creutzfeldt-Jakob disease (vCJD), and chronic wasting disease (CWD) – were specified in the investigation. The results of this investigation are published in the June issue of the Journal of the American Dietetic Association.
“While prion diseases are rare, they are generally fatal for anyone who becomes infected. More than anything else, the results of this study support the need for continued surveillance of prion diseases,” commented lead investigator Joseph Y. Abrams, MPH, National Center for Emerging and Zoonotic Infectious Diseases, CDC, Atlanta.”But it’s also important that people know the facts about these diseases, especially since this study shows that a good number of people have participated in activities that may expose them to infection-causing agents.”
Although rare, human prion diseases such as CJD may be related to BSE. Prion (proteinaceous infectious particles) diseases are a group of rare brain diseases that affect humans and animals. When a person gets a prion disease, brain function is impaired. This causes memory and personality changes, dementia, and problems with movement. All of these worsen over time. These diseases are invariably fatal. Since these diseases may take years to manifest, knowing the extent of human exposure to possible prion diseases could become important in the event of an outbreak.
CDC investigators evaluated the results of the 2006-2007 population survey conducted by the Foodborne Diseases Active Surveillance Network (FoodNet). This survey collects information on food consumption practices, health outcomes, and demographic characteristics of residents of the participating Emerging Infections Program sites. The survey was conducted in Connecticut, Georgia, Maryland, Minnesota, New Mexico, Oregon, and Tennessee, as well as five counties in the San Francisco Bay area, seven counties in the Greater Denver area, and 34 counties in western and northeastern New York.
Survey participants were asked about behaviors that could be associated with exposure to the agents causing BSE and CWD, including travel to the nine countries considered to be BSE-endemic (United Kingdom, Republic of Ireland, France, Portugal, Switzerland, Italy, the Netherlands, Germany, Spain) and the cumulative length of stay in each of those countries. Respondents were asked if they ever had hunted for deer or elk, and if that hunting had taken place in areas considered to be CWD-endemic (northeastern Colorado, southeastern Wyoming or southwestern Nebraska). They were also asked if they had ever consumed venison, the frequency of consumption, and whether the meat came from the wild.
The proportion of survey respondents who reported travel to at least one of the nine BSE endemic countries since 1980 was 29.5%. Travel to the United Kingdom was reported by 19.4% of respondents, higher than to any other BSE-endemic country. Among those who traveled, the median duration of travel to the United Kingdom (14 days) was longer than that of any other BSE-endemic country. Travelers to the UK were more likely to have spent at least 30 days in the country (24.9%) compared to travelers to any other BSE endemic country. The prevalence and extent of travel to the UK indicate that health concerns in the UK may also become issues for US residents.
The proportion of survey respondents reporting having hunted for deer or elk was 18.5% and 1.2% reported having hunted for deer or elk in CWD-endemic areas. Venison consumption was reported by 67.4% of FoodNet respondents, and 88.6% of those reporting venison consumption had obtained all of their meat from the wild. These findings reinforce the importance of CWD surveillance and control programs for wild deer and elk to reduce human exposure to the CWD agent. Hunters in CWD-endemic areas are advised to take simple precautions such as: avoiding consuming meat from sickly deer or elk, avoiding consuming brain or spinal cord tissues, minimizing the handling of brain and spinal cord tissues, and wearing gloves when field-dressing carcasses.
According to Abrams, “The 2006-2007 FoodNet population survey provides useful information should foodborne prion infection become an increasing public health concern in the future. The data presented describe the prevalence of important behaviors and their associations with demographic characteristics. Surveillance of BSE, CWD, and human prion diseases are critical aspects of addressing the burden of these diseases in animal populations and how that may relate to human health.”
###
The article is “Travel history, hunting, and venison consumption related to prion disease exposure, 2006-2007 FoodNet population survey” by Joseph Y. Abrams, MPH; Ryan A. Maddox, MPH; Alexis R Harvey, MPH; Lawrence B. Schonberger, MD; and Ermias D. Belay, MD. It appears in the Journal of the American Dietetic Association, Volume 111, Issue 6 (June 2011) published by Elsevier.
In an accompanying podcast CDC’s Joseph Y. Abrams discusses travel, hunting, and eating venison in relation to prion diseases. It is available at http://adajournal.org/content/podcast.
http://www.eurekalert.org/pub_releases/2011-05/ehs-cap051811.php
Thursday, May 26, 2011
Travel History, Hunting, and Venison Consumption Related to Prion Disease Exposure, 2006-2007 FoodNet Population Survey
Journal of the American Dietetic Association Volume 111, Issue 6 , Pages 858-863, June 2011.
Travel History, Hunting, and Venison Consumption Related to Prion Disease Exposure, 2006-2007 FoodNet Population Survey
Joseph Y. Abrams, MPH, Ryan A. Maddox, MPH , Alexis R. Harvey, MPH , Lawrence B. Schonberger, MD , Ermias D. Belay, MD
Accepted 15 November 2010. Abstract Full Text PDF References .
Abstract
The transmission of bovine spongiform encephalopathy (BSE) to human beings and the spread of chronic wasting disease (CWD) among cervids have prompted concerns about zoonotic transmission of prion diseases. Travel to the United Kingdom and other European countries, hunting for deer or elk, and venison consumption could result in the exposure of US residents to the agents that cause BSE and CWD. The Foodborne Diseases Active Surveillance Network 2006-2007 population survey was used to assess the prevalence of these behaviors among residents of 10 catchment areas across the United States. Of 17,372 survey respondents, 19.4% reported travel to the United Kingdom since 1980, and 29.5% reported travel to any of the nine European countries considered to be BSE-endemic since 1980. The proportion of respondents who had ever hunted deer or elk was 18.5%, and 1.2% had hunted deer or elk in a CWD–endemic area. More than two thirds (67.4%) reported having ever eaten deer or elk meat. Respondents who traveled spent more time in the United Kingdom (median 14 days) than in any other BSE-endemic country. Of the 11,635 respondents who had consumed venison, 59.8% ate venison at most one to two times during their year of highest consumption, and 88.6% had obtained all of their meat from the wild. The survey results were useful in determining the prevalence and frequency of behaviors that could be important factors for foodborne prion transmission.
http://www.adajournal.org/article/S0002-8223(11)00278-1/abstract
PLUS, THE CDC DID NOT PUT THIS WARNING OUT FOR THE WELL BEING OF THE DEER AND ELK ;
Thursday, May 26, 2011
Travel History, Hunting, and Venison Consumption Related to Prion Disease Exposure, 2006-2007 FoodNet Population Survey
Journal of the American Dietetic Association Volume 111, Issue 6 , Pages 858-863, June 2011.
http://transmissiblespongiformencephalopathy.blogspot.com/2011/05/travel-history-hunting-and-venison.html
NOR IS THE FDA recalling this CWD positive elk meat for the well being of the dead elk ;
Wednesday, March 18, 2009
Noah's Ark Holding, LLC, Dawson, MN RECALL Elk products contain meat derived from an elk confirmed to have CWD NV, CA, TX, CO, NY, UT, FL, OK RECALLS AND FIELD CORRECTIONS: FOODS CLASS II
http://chronic-wasting-disease.blogspot.com/2009/03/noahs-ark-holding-llc-dawson-mn-recall.html
Wednesday, March 18, 2009
Noah's Ark Holding, LLC, Dawson, MN RECALL Elk products contain meat derived from an elk confirmed to have CWD NV, CA, TX, CO, NY, UT, FL, OK RECALLS AND FIELD CORRECTIONS: FOODS CLASS II
http://chronic-wasting-disease.blogspot.com/2009/03/noahs-ark-holding-llc-dawson-mn-recall.html
Transmissible Spongiform Encephalopathies
Spongiform Encephalopathy in Captive Wild ZOO BSE INQUIRY
https://web.archive.org/web/20090506001201/http://www.bseinquiry.gov.uk/files/mb/m09a/tab03.pdf
BSE INQUIRY
CJD9/10022
October 1994
Mr R.N. Elmhirst Chairman British Deer Farmers Association Holly Lodge Spencers Lane
BerksWell Coventry CV7 7BZ
Dear Mr Elmhirst,
CREUTZFELDT-JAKOB DISEASE (CJD) SURVEILLANCE UNIT REPORT
Thank you for your recent letter concerning the publication of the third annual report from the CJD Surveillance Unit. I am sorry that you are dissatisfied with the way in which this report was published.
The Surveillance Unit is a completely independant outside body and the Department of Health is committed to publishing their reports as soon as they become available. In the circumstances it is not the practice to circulate the report for comment since the findings of the report would not be amended.. In future we can ensure that the British Deer Farmers Association receives a copy of the report in advance of publication.
The Chief Medical Officer has undertaken to keep the public fully informed of the results of any research in respect of CJD. This report was entirely the work of the unit and was produced completely independantly of the the Department.
The statistical results reqarding the consumption of venison was put into perspective in the body of the report and was not mentioned at all in the press release. Media attention regarding this report was low key but gave a realistic presentation of the statistical findings of the Unit. This approach to publication was successful in that consumption of venison was highlighted only once by the media ie. in the News at one television proqramme.
I believe that a further statement about the report, or indeed statistical links between CJD and consumption of venison, would increase, and quite possibly give damaging credence, to the whole issue. From the low key media reports of which I am aware it seems unlikely that venison consumption will suffer adversely, if at all.
http://web.archive.org/web/20030511010117/http://www.bseinquiry.gov.uk/files/yb/1994/10/00003001.pdf
*** The association between venison eating and risk of CJD shows similar pattern, with regular venison eating associated with a 9 FOLD INCREASE IN RISK OF CJD (p = 0.04). ***
*** The association between venison eating and risk of CJD shows similar pattern, with regular venison eating associated with a 9 FOLD INCREASE IN RISK OF CJD (p = 0.04). ***
*** The association between venison eating and risk of CJD shows similar pattern, with regular venison eating associated with a 9 FOLD INCREASE IN RISK OF CJD (p = 0.04). ***
There is some evidence that risk of CJD INCREASES WITH INCREASING FREQUENCY OF LAMB EATING (p = 0.02).
The evidence for such an association between beef eating and CJD is weaker (p = 0.14). When only controls for whom a relative was interviewed are included, this evidence becomes a little STRONGER (p = 0.08).
snip...
It was found that when veal was included in the model with another exposure, the association between veal and CJD remained statistically significant (p = < 0.05 for all exposures), while the other exposures ceased to be statistically significant (p = > 0.05).
snip...
In conclusion, an analysis of dietary histories revealed statistical associations between various meats/animal products and INCREASED RISK OF CJD. When some account was taken of possible confounding, the association between VEAL EATING AND RISK OF CJD EMERGED AS THE STRONGEST OF THESE ASSOCIATIONS STATISTICALLY. ...
snip...
In the study in the USA, a range of foodstuffs were associated with an increased risk of CJD, including liver consumption which was associated with an apparent SIX-FOLD INCREASE IN THE RISK OF CJD. By comparing the data from 3 studies in relation to this particular dietary factor, the risk of liver consumption became non-significant with an odds ratio of 1.2 (PERSONAL COMMUNICATION, PROFESSOR A. HOFMAN. ERASMUS UNIVERSITY, ROTTERDAM). (???...TSS)
snip...see full report ;
https://web.archive.org/web/20170126073306/http://collections..europarchive..org/tna/20090505194948/http://bseinquiry.gov.uk/files/yb/1994/08/00004001.pdf
ALSO, I FIND THIS VERY DISTURBING...SEE;
***> Prion 2018 P74 High Prevalence of CWD prions in male reproductive samples
Carlos Kramm (1,2), Ruben Gomez-Gutierrez (1,3), Tracy Nichols (4), Claudio Soto (1) and Rodrigo Morales (1)
READING OVER THE PRION 2018 ABSTRACT BOOK, LOOKS LIKE THEY FOUND THAT Protein Misfolding Cyclic Amplification PMCA results showed positive CWD prion detection in testes, epididymis and seminal fluid samples. seems also the scientists are worried about any potential mechanisms of CWD spreading and they want to decrease putative interindividual transmission associated to insemination using CWD contaminated specimens, if that might occur under natural conditions. i have been concerned about this for some time with BSE super-ovulation and since;
PrPSc detection and infectivity in semen from scrapie-infected sheep
PITUITARY EXTRACT
This was used to help cows super ovulate. This tissue was considered to be of greatest risk of containing BSE and consequently transmitting the disease...
This was used to help cows super ovulate. This tissue was considered to be of greatest risk of containing BSE and consequently transmitting the disease...
MANAGEMENT IN CONFIDENCE
CERTIFIED BSE-FREE HERDS FOR SOURCE OF MATERIAL FOR BIOLOGICAL PRODUCTS
Tuesday, February 8, 2011
U.S.A. 50 STATE BSE MAD COW CONFERENCE CALL Jan. 9, 2001
HAVE YOU BEEN THUNDERSTRUCK?
SUNDAY, AUGUST 02, 2015
TEXAS CWD, Have you been ThunderStruck, deer semen, straw bred bucks, super ovulation, and the potential TSE Prion connection, what if?
Court papers state that in February 2007, Favero acquired 184 straws of whitetail deer semen valued at about $92,000 from a buck named “Diablo'” that he knew had been illegally taken out of Texas, and again in January 2008 took another 110 straws of semen from a buck named “Thunderstruck.” (Read more in the court paper posted at the bottom of this entry.)
SUNDAY, AUGUST 19, 2018
Texas Deer Farms, Growing Freakish Antlers, and CWD TSE Prion aka mad deer disease
FRIDAY, MARCH 30, 2018
Docket No. APHIS-2018-0011 Chronic Wasting Disease Herd Certification Program Standards Singeltary Submission March 30, 2018
Terry S. Singeltary Sr., Bacliff, Texas USA 77518 flounder9@verizon....net
Attachments (1) Docket No. APHIS-2018-0011 Chronic Wasting Disease Herd Certification Program Standards Singeltary View Attachment:View as format pdf
Friday, December 14, 2012
DEFRA U.K. What is the risk of Chronic Wasting Disease CWD being introduced into Great Britain? A Qualitative Risk Assessment October 2012
snip.....
In the USA, under the Food and Drug Administration's BSE Feed Regulation (21 CFR 589.2000) most material (exceptions include milk, tallow, and gelatin) from deer and elk is prohibited for use in feed for ruminant animals. With regards to feed for non-ruminant animals, under FDA law, CWD positive deer may not be used for any animal feed or feed ingredients. For elk and deer considered at high risk for CWD, the FDA recommends that these animals do not enter the animal feed system. However, this recommendation is guidance and not a requirement by law.
Animals considered at high risk for CWD include:
1) animals from areas declared to be endemic for CWD and/or to be CWD eradication zones and
2) deer and elk that at some time during the 60-month period prior to slaughter were in a captive herd that contained a CWD-positive animal.
Therefore, in the USA, materials from cervids other than CWD positive animals may be used in animal feed and feed ingredients for non-ruminants.
The amount of animal PAP that is of deer and/or elk origin imported from the USA to GB can not be determined, however, as it is not specified in TRACES. It may constitute a small percentage of the 8412 kilos of non-fish origin processed animal proteins that were imported from US into GB in 2011.
Overall, therefore, it is considered there is a __greater than negligible risk___ that (nonruminant) animal feed and pet food containing deer and/or elk protein is imported into GB.
There is uncertainty associated with this estimate given the lack of data on the amount of deer and/or elk protein possibly being imported in these products.
snip.....
36% in 2007 (Almberg et al., 2011). In such areas, population declines of deer of up to 30 to 50% have been observed (Almberg et al., 2011). In areas of Colorado, the prevalence can be as high as 30% (EFSA, 2011).
The clinical signs of CWD in affected adults are weight loss and behavioural changes that can span weeks or months (Williams, 2005). In addition, signs might include excessive salivation, behavioural alterations including a fixed stare and changes in interaction with other animals in the herd, and an altered stance (Williams, 2005). These signs are indistinguishable from cervids experimentally infected with bovine spongiform encephalopathy (BSE).
Given this, if CWD was to be introduced into countries with BSE such as GB, for example, infected deer populations would need to be tested to differentiate if they were infected with CWD or BSE to minimise the risk of BSE entering the human food-chain via affected venison.
snip.....
The rate of transmission of CWD has been reported to be as high as 30% and can approach 100% among captive animals in endemic areas (Safar et al., 2008).
snip.....
In summary, in endemic areas, there is a medium probability that the soil and surrounding environment is contaminated with CWD prions and in a bioavailable form. In rural areas where CWD has not been reported and deer are present, there is a greater than negligible risk the soil is contaminated with CWD prion.
snip.....
In summary, given the volume of tourists, hunters and servicemen moving between GB and North America, the probability of at least one person travelling to/from a CWD affected area and, in doing so, contaminating their clothing, footwear and/or equipment prior to arriving in GB is greater than negligible... For deer hunters, specifically, the risk is likely to be greater given the increased contact with deer and their environment. However, there is significant uncertainty associated with these estimates.
snip.....
Therefore, it is considered that farmed and park deer may have a higher probability of exposure to CWD transferred to the environment than wild deer given the restricted habitat range and higher frequency of contact with tourists and returning GB residents.
snip.....
TUESDAY, APRIL 18, 2017
*** EXTREME USA FDA PART 589 TSE PRION FEED LOOP HOLE STILL EXIST, AND PRICE OF POKER GOES UP ***
TUESDAY, JANUARY 17, 2017
FDA PART 589 -- SUBSTANCES PROHIBITED FROM USE IN ANIMAL FOOD OR FEEDVIOLATIONS OFFICIAL ACTION INDICATED OAI UPDATE 2016 to 2017 BSE TSE PRION
THIS April, 4, 2017
violation of the mad cow 21 CFR 589.2000 OAI is very serious for the great state of Michigan, some 20 years post FDA mad cow feed of August 1997. if would most likely take a FOIA request and a decade of wrangling to find out more.
TUESDAY, JANUARY 17, 2017
FDA PART 589 -- SUBSTANCES PROHIBITED FROM USE IN ANIMAL FOOD OR FEEDVIOLATIONS OFFICIAL ACTION INDICATED OAI UPDATE 2016 to 2017 BSE TSE PRION
FDA PART 589 -- SUBSTANCES PROHIBITED FROM USE IN ANIMAL FOOD OR FEEDVIOLATIONS OFFICIAL ACTION INDICATED OAI UPDATE 2016 to 2017 BSE TSE PRION
I would kindly like to comment on this FDA BSE/Ruminant Feed Inspections Firms Inventory (excel format)4 format, for reporting these breaches of BSE TSE prion protocols, from the extensive mad cow feed ban warning letters the fda use to put out for each violations. simply put, this excel format sucks, and the FDA et al intentionally made it this difficult to follow the usda fda mad cow follies. this is an intentional format to make it as difficult as possible to follow these breaches of the mad cow TSE prion safety feed protocols. to have absolutely no chronological or numerical order, and to format such violations in a way that they are almost impossible to find, says a lot about just how far the FDA and our fine federal friends will go through to hide these continued violations of the BSE TSE prion mad cow feed ban, and any breaches of protocols there from. once again, the wolf guarding the henhouse $$$
NAI = NO ACTION INDICATED
OAI = OFFICIAL ACTION INDICATED
VAI = VOLUNTARY ACTION INDICATED
RTS = REFERRED TO STATE
OAI (Official Action Indicated) when inspectors find significant objectionable conditions or practices and believe that regulatory sanctions are warranted to address the establishment’s lack of compliance with the regulation. An example of an OAI classification would be findings of manufacturing procedures insufficient to ensure that ruminant feed is not contaminated with prohibited material. Inspectors will promptly re-inspect facilities classified OAI after regulatory sanctions have been applied to determine whether the corrective actions are adequate to address the objectionable conditions.
2016
ONE more thing, please remember, the label does not have to say ''deer ration'' for cervid to be pumped up with. you can get the same ''high protein'' from many sources of high protein feed for animals other than cattle, and feed them to cervid...
Saturday, August 29, 2009
FOIA REQUEST FEED RECALL 2009 Product may have contained prohibited materials Bulk Whole Barley, Recall # V-256-2009
Friday, September 4, 2009
FOIA REQUEST ON FEED RECALL PRODUCT 429,128 lbs. feed for ruminant animals may have been contaminated with prohibited material Recall # V-258-2009
WEDNESDAY, JULY 11, 2018
CONFIDENTIAL IN CONFIDENCE SPONGIFORM ENCEPHALOPATHY OF PIGS FDA EMERGENCY REQUEST FOR RULE CHANGE USA Section 21 C.F.R. 589.2000
TUESDAY, JULY 10, 2018
CONFIDENTIAL IN CONFIDENCE SPONGIFORM ENCEPHALOPATHY OF PIGS
*** ''but feeding of other ruminant protein, including scrapie-infected sheep, can continue to pigs.''
CONFIDENTIAL SPONGIFORM ENCEPHALOPATHY OF PIGS
O.05: Transmission of prions to primates after extended silent incubation periods: Implications for BSE and scrapie risk assessment in human populations
Emmanuel Comoy, Jacqueline Mikol, Valerie Durand, Sophie Luccantoni, Evelyne Correia, Nathalie Lescoutra, Capucine Dehen, and Jean-Philippe Deslys Atomic Energy Commission; Fontenay-aux-Roses, France
Prion diseases (PD) are the unique neurodegenerative proteinopathies reputed to be transmissible under field conditions since decades. The transmission of Bovine Spongiform Encephalopathy (BSE) to humans evidenced that an animal PD might be zoonotic under appropriate conditions. Contrarily, in the absence of obvious (epidemiological or experimental) elements supporting a transmission or genetic predispositions, PD, like the other proteinopathies, are reputed to occur spontaneously (atpical animal prion strains, sporadic CJD summing 80% of human prion cases).
Non-human primate models provided the first evidences supporting the transmissibiity of human prion strains and the zoonotic potential of BSE. Among them, cynomolgus macaques brought major information for BSE risk assessment for human health (Chen, 2014), according to their phylogenetic proximity to humans and extended lifetime. We used this model to assess the zoonotic potential of other animal PD from bovine, ovine and cervid origins even after very long silent incubation periods.
*** We recently observed the direct transmission of a natural classical scrapie isolate to macaque after a 10-year silent incubation period,
***with features similar to some reported for human cases of sporadic CJD, albeit requiring fourfold long incubation than BSE. Scrapie, as recently evoked in humanized mice (Cassard, 2014),
***is the third potentially zoonotic PD (with BSE and L-type BSE),
***thus questioning the origin of human sporadic cases.
We will present an updated panorama of our different transmission studies and discuss the implications of such extended incubation periods on risk assessment of animal PD for human health.
===============
***thus questioning the origin of human sporadic cases***
===============
***our findings suggest that possible transmission risk of H-type BSE to sheep and human. Bioassay will be required to determine whether the PMCA products are infectious to these animals.
==============
https://prion2015.files.wordpress.com/2015/05/prion2015abstracts.pdf
***Transmission data also revealed that several scrapie prions propagate in HuPrP-Tg mice with efficiency comparable to that of cattle BSE. While the efficiency of transmission at primary passage was low, subsequent passages resulted in a highly virulent prion disease in both Met129 and Val129 mice.
***Transmission of the different scrapie isolates in these mice leads to the emergence of prion strain phenotypes that showed similar characteristics to those displayed by MM1 or VV2 sCJD prion.
***These results demonstrate that scrapie prions have a zoonotic potential and raise new questions about the possible link between animal and human prions.
http://www.tandfonline.com/doi/abs/10.1080/19336896.2016.1163048?journalCode=kprn20
PRION 2016 TOKYO
Saturday, April 23, 2016
SCRAPIE WS-01: Prion diseases in animals and zoonotic potential 2016
Prion. 10:S15-S21. 2016 ISSN: 1933-6896 printl 1933-690X online
Taylor & Francis
Prion 2016 Animal Prion Disease Workshop Abstracts
WS-01: Prion diseases in animals and zoonotic potential
Juan Maria Torres a, Olivier Andreoletti b, J uan-Carlos Espinosa a. Vincent Beringue c. Patricia Aguilar a,
Natalia Fernandez-Borges a. and Alba Marin-Moreno a
"Centro de Investigacion en Sanidad Animal ( CISA-INIA ). Valdeolmos, Madrid. Spain; b UMR INRA -ENVT 1225 Interactions Holes Agents Pathogenes. ENVT. Toulouse. France: "UR892. Virologie lmmunologie MolécuIaires, Jouy-en-Josas. France
Dietary exposure to bovine spongiform encephalopathy (BSE) contaminated bovine tissues is considered as the origin of variant Creutzfeldt Jakob (vCJD) disease in human. To date, BSE agent is the only recognized zoonotic prion.. Despite the variety of Transmissible Spongiform Encephalopathy (TSE) agents that have been circulating for centuries in farmed ruminants there is no apparent epidemiological link between exposure to ruminant products and the occurrence of other form of TSE in human like sporadic Creutzfeldt Jakob Disease (sCJD). However, the zoonotic potential of the diversity of circulating TSE agents has never been systematically assessed. The major issue in experimental assessment of TSEs zoonotic potential lies in the modeling of the ‘species barrier‘, the biological phenomenon that limits TSE agents’ propagation from a species to another. In the last decade, mice genetically engineered to express normal forms of the human prion protein has proved essential in studying human prions pathogenesis and modeling the capacity of TSEs to cross the human species barrier.
To assess the zoonotic potential of prions circulating in farmed ruminants, we study their transmission ability in transgenic mice expressing human PrPC (HuPrP-Tg). Two lines of mice expressing different forms of the human PrPC (129Met or 129Val) are used to determine the role of the Met129Val dimorphism in susceptibility/resistance to the different agents.
These transmission experiments confirm the ability of BSE prions to propagate in 129M- HuPrP-Tg mice and demonstrate that Met129 homozygotes may be susceptible to BSE in sheep or goat to a greater degree than the BSE agent in cattle and that these agents can convey molecular properties and neuropathological indistinguishable from vCJD. However homozygous 129V mice are resistant to all tested BSE derived prions independently of the originating species suggesting a higher transmission barrier for 129V-PrP variant.
Transmission data also revealed that several scrapie prions propagate in HuPrP-Tg mice with efficiency comparable to that of cattle BSE. While the efficiency of transmission at primary passage was low, subsequent passages resulted in a highly virulent prion disease in both Met129 and Val129 mice.
Transmission of the different scrapie isolates in these mice leads to the emergence of prion strain phenotypes that showed similar characteristics to those displayed by MM1 or VV2 sCJD prion.
These results demonstrate that scrapie prions have a zoonotic potential and raise new questions about the possible link between animal and human prions.
http://www.tandfonline.com/doi/abs/10.1080/19336896.2016.1163048?journalCode=kprn20
why do we not want to do TSE transmission studies on chimpanzees $
5. A positive result from a chimpanzee challenged severly would likely create alarm in some circles even if the result could not be interpreted for man. I have a view that all these agents could be transmitted provided a large enough dose by appropriate routes was given and the animals kept long enough. Until the mechanisms of the species barrier are more clearly understood it might be best to retain that hypothesis.
snip...
R. BRADLEY
https://web.archive.org/web/20170126051158/http://collections.europarchive.org/tna/20080102222950/http://www.bseinquiry.gov.uk/files/yb/1990/09/23001001.pdf
Title: Transmission of scrapie prions to primate after an extended silent incubation period)
*** In complement to the recent demonstration that humanized mice are susceptible to scrapie, we report here the first observation of direct transmission of a natural classical scrapie isolate to a macaque after a 10-year incubation period. Neuropathologic examination revealed all of the features of a prion disease: spongiform change, neuronal loss, and accumulation of PrPres throughout the CNS.
*** This observation strengthens the questioning of the harmlessness of scrapie to humans, at a time when protective measures for human and animal health are being dismantled and reduced as c-BSE is considered controlled and being eradicated.
*** Our results underscore the importance of precautionary and protective measures and the necessity for long-term experimental transmission studies to assess the zoonotic potential of other animal prion strains.
http://www.ars.usda.gov/research/publications/publications.htm?SEQ_NO_115=313160
***> Moreover, sporadic disease has never been observed in breeding colonies or primate research laboratories, most notably among hundreds of animals over several decades of study at the National Institutes of Health25, and in nearly twenty older animals continuously housed in our own facility. <***
Transmission of scrapie prions to primate after an extended silent incubation period
Emmanuel E. Comoy, Jacqueline Mikol, Sophie Luccantoni-Freire, Evelyne Correia, Nathalie Lescoutra-Etchegaray, Valérie Durand, Capucine Dehen, Olivier Andreoletti, Cristina Casalone, Juergen A. Richt, Justin J. Greenlee, Thierry Baron, Sylvie L. Benestad, Paul Brown & Jean-Philippe Deslys Scientific Reports volume 5, Article number: 11573 (2015) | Download Citation
Abstract
Classical bovine spongiform encephalopathy (c-BSE) is the only animal prion disease reputed to be zoonotic, causing variant Creutzfeldt-Jakob disease (vCJD) in humans and having guided protective measures for animal and human health against animal prion diseases. Recently, partial transmissions to humanized mice showed that the zoonotic potential of scrapie might be similar to c-BSE. We here report the direct transmission of a natural classical scrapie isolate to cynomolgus macaque, a highly relevant model for human prion diseases, after a 10-year silent incubation period, with features similar to those reported for human cases of sporadic CJD. Scrapie is thus actually transmissible to primates with incubation periods compatible with their life expectancy, although fourfold longer than BSE. Long-term experimental transmission studies are necessary to better assess the zoonotic potential of other prion diseases with high prevalence, notably Chronic Wasting Disease of deer and elk and atypical/Nor98 scrapie.
SNIP...
Discussion We describe the transmission of spongiform encephalopathy in a non-human primate inoculated 10 years earlier with a strain of sheep c-scrapie. Because of this extended incubation period in a facility in which other prion diseases are under study, we are obliged to consider two alternative possibilities that might explain its occurrence. We first considered the possibility of a sporadic origin (like CJD in humans). Such an event is extremely improbable because the inoculated animal was 14 years old when the clinical signs appeared, i.e. about 40% through the expected natural lifetime of this species, compared to a peak age incidence of 60–65 years in human sporadic CJD, or about 80% through their expected lifetimes. Moreover, sporadic disease has never been observed in breeding colonies or primate research laboratories, most notably among hundreds of animals over several decades of study at the National Institutes of Health25, and in nearly twenty older animals continuously housed in our own facility.
The second possibility is a laboratory cross-contamination. Three facts make this possibility equally unlikely. First, handling of specimens in our laboratory is performed with fastidious attention to the avoidance of any such cross-contamination. Second, no laboratory cross-contamination has ever been documented in other primate laboratories, including the NIH, even between infected and uninfected animals housed in the same or adjacent cages with daily intimate contact (P. Brown, personal communication). Third, the cerebral lesion profile is different from all the other prion diseases we have studied in this model19, with a correlation between cerebellar lesions (massive spongiform change of Purkinje cells, intense PrPres staining and reactive gliosis26) and ataxia. The iron deposits present in the globus pallidus are a non specific finding that have been reported previously in neurodegenerative diseases and aging27. Conversely, the thalamic lesion was reminiscent of a metabolic disease due to thiamine deficiency28 but blood thiamine levels were within normal limits (data not shown). The preferential distribution of spongiform change in cortex associated with a limited distribution in the brainstem is reminiscent of the lesion profile in MM2c and VV1 sCJD patients29, but interspecies comparison of lesion profiles should be interpreted with caution. It is of note that the same classical scrapie isolate induced TSE in C57Bl/6 mice with similar incubation periods and lesional profiles as a sample derived from a MM1 sCJD patient30.
We are therefore confident that the illness in this cynomolgus macaque represents a true transmission of a sheep c-scrapie isolate directly to an old-world monkey, which taxonomically resides in the primate subdivision (parvorder of catarrhini) that includes humans. With an homology of its PrP protein with humans of 96.4%31, cynomolgus macaque constitutes a highly relevant model for assessing zoonotic risk of prion diseases. Since our initial aim was to show the absence of transmission of scrapie to macaques in the worst-case scenario, we obtained materials from a flock of naturally-infected sheep, affecting animals with different genotypes32. This c-scrapie isolate exhibited complete transmission in ARQ/ARQ sheep (332 ± 56 days) and Tg338 transgenic mice expressing ovine VRQ/VRQ prion protein (220 ± 5 days) (O. Andreoletti, personal communication). From the standpoint of zoonotic risk, it is important to note that sheep with c-scrapie (including the isolate used in our study) have demonstrable infectivity throughout their lymphoreticular system early in the incubation period of the disease (3 months-old for all the lymphoid organs, and as early as 2 months-old in gut-associated lymph nodes)33. In addition, scrapie infectivity has been identified in blood34, milk35 and skeletal muscle36 from asymptomatic but scrapie infected small ruminants which implies a potential dietary exposure for consumers.
Two earlier studies have reported the occurrence of clinical TSE in cynomolgus macaques after exposures to scrapie isolates. In the first study, the “Compton” scrapie isolate (derived from an English sheep) and serially propagated for 9 passages in goats did not transmit TSE in cynomolgus macaque, rhesus macaque or chimpanzee within 7 years following intracerebral challenge1; conversely, after 8 supplementary passages in conventional mice, this “Compton” isolate induced TSE in a cynomolgus macaque 5 years after intracerebral challenge, but rhesus macaques and chimpanzee remained asymptomatic 8.5 years post-exposure8. However, multiple successive passages that are classically used to select laboratory-adapted prion strains can significantly modify the initial properties of a scrapie isolate, thus questioning the relevance of zoonotic potential for the initial sheep-derived isolate. The same isolate had also induced disease into squirrel monkeys (new-world monkey)9. A second historical observation reported that a cynomolgus macaque developed TSE 6 years post-inoculation with brain homogenate from a scrapie-infected Suffolk ewe (derived from USA), whereas a rhesus macaque and a chimpanzee exposed to the same inoculum remained healthy 9 years post-exposure1. This inoculum also induced TSE in squirrel monkeys after 4 passages in mice. Other scrapie transmission attempts in macaque failed but had more shorter periods of observation in comparison to the current study. Further, it is possible that there are differences in the zoonotic potential of different scrapie strains.
The most striking observation in our study is the extended incubation period of scrapie in the macaque model, which has several implications. Firstly, our observations constitute experimental evidence in favor of the zoonotic potential of c-scrapie, at least for this isolate that has been extensively studied32,33,34,35,36. The cross-species zoonotic ability of this isolate should be confirmed by performing duplicate intracerebral exposures and assessing the transmissibility by the oral route (a successful transmission of prion strains through the intracerebral route may not necessarily indicate the potential for oral transmission37). However, such confirmatory experiments may require more than one decade, which is hardly compatible with current general management and support of scientific projects; thus this study should be rather considered as a case report.
Secondly, transmission of c-BSE to primates occurred within 8 years post exposure for the lowest doses able to transmit the disease (the survival period after inoculation is inversely proportional to the initial amount of infectious inoculum). The occurrence of scrapie 10 years after exposure to a high dose (25 mg) of scrapie-infected sheep brain suggests that the macaque has a higher species barrier for sheep c-scrapie than c-BSE, although it is notable that previous studies based on in vitro conversion of PrP suggested that BSE and scrapie prions would have a similar conversion potential for human PrP38.
Thirdly, prion diseases typically have longer incubation periods after oral exposure than after intracerebral inoculations: since humans can develop Kuru 47 years after oral exposure39, an incubation time of several decades after oral exposure to scrapie would therefore be expected, leading the disease to occur in older adults, i.e. the peak age for cases considered to be sporadic disease, and making a distinction between scrapie-associated and truly sporadic disease extremely difficult to appreciate.
Fourthly, epidemiologic evidence is necessary to confirm the zoonotic potential of an animal disease suggested by experimental studies. A relatively short incubation period and a peculiar epidemiological situation (e.g., all the first vCJD cases occurring in the country with the most important ongoing c-BSE epizootic) led to a high degree of suspicion that c-BSE was the cause of vCJD. Sporadic CJD are considered spontaneous diseases with an almost stable and constant worldwide prevalence (0.5–2 cases per million inhabitants per year), and previous epidemiological studies were unable to draw a link between sCJD and classical scrapie6,7,40,41, even though external causes were hypothesized to explain the occurrence of some sCJD clusters42,43,44. However, extended incubation periods exceeding several decades would impair the predictive values of epidemiological surveillance for prion diseases, already weakened by a limited prevalence of prion diseases and the multiplicity of isolates gathered under the phenotypes of “scrapie” and “sporadic CJD”.
Fifthly, considering this 10 year-long incubation period, together with both laboratory and epidemiological evidence of decade or longer intervals between infection and clinical onset of disease, no premature conclusions should be drawn from negative transmission studies in cynomolgus macaques with less than a decade of observation, as in the aforementioned historical transmission studies of scrapie to primates1,8,9. Our observations and those of others45,46 to date are unable to provide definitive evidence regarding the zoonotic potential of CWD, atypical/Nor98 scrapie or H-type BSE. The extended incubation period of the scrapie-affected macaque in the current study also underscores the limitations of rodent models expressing human PrP for assessing the zoonotic potential of some prion diseases since their lifespan remains limited to approximately two years21,47,48. This point is illustrated by the fact that the recently reported transmission of scrapie to humanized mice was not associated with clinical signs for up to 750 days and occurred in an extreme minority of mice with only a marginal increase in attack rate upon second passage13. The low attack rate in these studies is certainly linked to the limited lifespan of mice compared to the very long periods of observation necessary to demonstrate the development of scrapie. Alternatively, one could estimate that a successful second passage is the result of strain adaptation to the species barrier, thus poorly relevant of the real zoonotic potential of the original scrapie isolate of sheep origin49. The development of scrapie in this primate after an incubation period compatible with its lifespan complements the study conducted in transgenic (humanized) mice; taken together these studies suggest that some isolates of sheep scrapie can promote misfolding of the human prion protein and that scrapie can develop within the lifespan of some primate species.
In addition to previous studies on scrapie transmission to primate1,8,9 and the recently published study on transgenic humanized mice13, our results constitute new evidence for recommending that the potential risk of scrapie for human health should not be dismissed. Indeed, human PrP transgenic mice and primates are the most relevant models for investigating the human transmission barrier. To what extent such models are informative for measuring the zoonotic potential of an animal TSE under field exposure conditions is unknown. During the past decades, many protective measures have been successfully implemented to protect cattle from the spread of c-BSE, and some of these measures have been extended to sheep and goats to protect from scrapie according to the principle of precaution. Since cases of c-BSE have greatly reduced in number, those protective measures are currently being challenged and relaxed in the absence of other known zoonotic animal prion disease. We recommend that risk managers should be aware of the long term potential risk to human health of at least certain scrapie isolates, notably for lymphotropic strains like the classical scrapie strain used in the current study. Relatively high amounts of infectivity in peripheral lymphoid organs in animals infected with these strains could lead to contamination of food products produced for human consumption. Efforts should also be maintained to further assess the zoonotic potential of other animal prion strains in long-term studies, notably lymphotropic strains with high prevalence like CWD, which is spreading across North America, and atypical/Nor98 scrapie (Nor98)50 that was first detected in the past two decades and now represents approximately half of all reported cases of prion diseases in small ruminants worldwide, including territories previously considered as scrapie free.. Even if the prevailing view is that sporadic CJD is due to the spontaneous formation of CJD prions, it remains possible that its apparent sporadic nature may, at least in part, result from our limited capacity to identify an environmental origin.
Singeltary on Scrapie and human transmission way back, see;
***> CONGRESSIONAL ABSTRACTS PRION CONFERENCE 2018
P69 Experimental transmission of CWD from white-tailed deer to co-housed reindeer
Mitchell G (1), Walther I (1), Staskevicius A (1), Soutyrine A (1), Balachandran A (1)
(1) National & OIE Reference Laboratory for Scrapie and CWD, Canadian Food Inspection Agency, Ottawa, Ontario, Canada.
Chronic wasting disease (CWD) continues to be detected in wild and farmed cervid populations of North America, affecting predominantly white-tailed deer, mule deer and elk. Extensive herds of wild caribou exist in northern regions of Canada, although surveillance has not detected the presence of CWD in this population. Oral experimental transmission has demonstrated that reindeer, a species closely related to caribou, are susceptible to CWD. Recently, CWD was detected for the first time in Europe, in wild Norwegian reindeer, advancing the possibility that caribou in North America could also become infected. Given the potential overlap in habitat between wild CWD-infected cervids and wild caribou herds in Canada, we sought to investigate the horizontal transmissibility of CWD from white-tailed deer to reindeer.
Two white-tailed deer were orally inoculated with a brain homogenate prepared from a farmed Canadian white-tailed deer previously diagnosed with CWD. Two reindeer, with no history of exposure to CWD, were housed in the same enclosure as the white-tailed deer, 3.5 months after the deer were orally inoculated. The white-tailed deer developed clinical signs consistent with CWD beginning at 15.2 and 21 months post-inoculation (mpi), and were euthanized at 18.7 and 23.1 mpi, respectively. Confirmatory testing by immunohistochemistry (IHC) and western blot demonstrated widespread aggregates of pathological prion protein (PrPCWD) in the central nervous system and lymphoid tissues of both inoculated white-tailed deer. Both reindeer were subjected to recto-anal mucosal associated lymphoid tissue (RAMALT) biopsy at 20 months post-exposure (mpe) to the white-tailed deer. The biopsy from one reindeer contained PrPCWD confirmed by IHC. This reindeer displayed only subtle clinical evidence of disease prior to a rapid decline in condition requiring euthanasia at 22.5 mpe. Analysis of tissues from this reindeer by IHC revealed widespread PrPCWD deposition, predominantly in central nervous system and lymphoreticular tissues. Western blot molecular profiles were similar between both orally inoculated white-tailed deer and the CWD positive reindeer. Despite sharing the same enclosure, the other reindeer was RAMALT negative at 20 mpe, and PrPCWD was not detected in brainstem and lymphoid tissues following necropsy at 35 mpe. Sequencing of the prion protein gene from both reindeer revealed differences at several codons, which may have influenced susceptibility to infection.
Natural transmission of CWD occurs relatively efficiently amongst cervids, supporting the expanding geographic distribution of disease and the potential for transmission to previously naive populations. The efficient horizontal transmission of CWD from white-tailed deer to reindeer observed here highlights the potential for reindeer to become infected if exposed to other cervids or environments infected with CWD.
TITLE: PATHOLOGICAL FEATURES OF CHRONIC WASTING DISEASE IN REINDEER AND DEMONSTRATION OF HORIZONTAL TRANSMISSION
*** DECEMBER 2016 CDC EMERGING INFECTIOUS DISEASE JOURNAL CWD HORIZONTAL TRANSMISSION
Infectious agent of sheep scrapie may persist in the environment for at least 16 years
*** Nine of these recurrences occurred 14–21 years after culling
Gudmundur Georgsson,1 Sigurdur Sigurdarson2 and Paul Brown3
Correspondence Gudmundur Georgsson ggeorgs@hi.is
1 Institute for Experimental Pathology, University of Iceland, Keldur v/vesturlandsveg, IS-112 Reykjavı´k, Iceland
2 Laboratory of the Chief Veterinary Officer, Keldur, Iceland
3 Bethesda, Maryland, USA Received 7 March 2006 Accepted 6 August 2006
In 1978, a rigorous programme was implemented to stop the spread of, and subsequently eradicate, sheep scrapie in Iceland. Affected flocks were culled, premises were disinfected and, after 2–3 years, restocked with lambs from scrapie-free areas. Between 1978 and 2004, scrapie recurred on 33 farms. Nine of these recurrences occurred 14–21 years after culling, apparently as the result of environmental contamination, but outside entry could not always be absolutely excluded. Of special interest was one farm with a small, completely self-contained flock where scrapie recurred 18 years after culling, 2 years after some lambs had been housed in an old sheephouse that had never been disinfected. Epidemiological investigation established with near certitude that the disease had not been introduced from the outside and it is concluded that the agent may have persisted in the old sheep-house for at least 16 years.
*** Infectious agent of sheep scrapie may persist in the environment for at least 16 years ***
Gudmundur Georgsson1, Sigurdur Sigurdarson2 and Paul Brown3
Using in vitro Prion replication for high sensitive detection of prions and prionlike proteins and for understanding mechanisms of transmission.
Claudio Soto Mitchell Center for Alzheimer's diseases and related Brain disorders, Department of Neurology, University of Texas Medical School at Houston.
Prion and prion-like proteins are misfolded protein aggregates with the ability to selfpropagate to spread disease between cells, organs and in some cases across individuals. I n T r a n s m i s s i b l e s p o n g i f o r m encephalopathies (TSEs), prions are mostly composed by a misfolded form of the prion protein (PrPSc), which propagates by transmitting its misfolding to the normal prion protein (PrPC). The availability of a procedure to replicate prions in the laboratory may be important to study the mechanism of prion and prion-like spreading and to develop high sensitive detection of small quantities of misfolded proteins in biological fluids, tissues and environmental samples. Protein Misfolding Cyclic Amplification (PMCA) is a simple, fast and efficient methodology to mimic prion replication in the test tube. PMCA is a platform technology that may enable amplification of any prion-like misfolded protein aggregating through a seeding/nucleation process. In TSEs, PMCA is able to detect the equivalent of one single molecule of infectious PrPSc and propagate prions that maintain high infectivity, strain properties and species specificity. Using PMCA we have been able to detect PrPSc in blood and urine of experimentally infected animals and humans affected by vCJD with high sensitivity and specificity. Recently, we have expanded the principles of PMCA to amplify amyloid-beta (Aβ) and alphasynuclein (α-syn) aggregates implicated in Alzheimer's and Parkinson's diseases, respectively. Experiments are ongoing to study the utility of this technology to detect Aβ and α-syn aggregates in samples of CSF and blood from patients affected by these diseases.
=========================
***>>> Recently, we have been using PMCA to study the role of environmental prion contamination on the horizontal spreading of TSEs. These experiments have focused on the study of the interaction of prions with plants and environmentally relevant surfaces. Our results show that plants (both leaves and roots) bind tightly to prions present in brain extracts and excreta (urine and feces) and retain even small quantities of PrPSc for long periods of time. Strikingly, ingestion of prioncontaminated leaves and roots produced disease with a 100% attack rate and an incubation period not substantially longer than feeding animals directly with scrapie brain homogenate. Furthermore, plants can uptake prions from contaminated soil and transport them to different parts of the plant tissue (stem and leaves). Similarly, prions bind tightly to a variety of environmentally relevant surfaces, including stones, wood, metals, plastic, glass, cement, etc. Prion contaminated surfaces efficiently transmit prion disease when these materials were directly injected into the brain of animals and strikingly when the contaminated surfaces were just placed in the animal cage. These findings demonstrate that environmental materials can efficiently bind infectious prions and act as carriers of infectivity, suggesting that they may play an important role in the horizontal transmission of the disease.
========================
Since its invention 13 years ago, PMCA has helped to answer fundamental questions of prion propagation and has broad applications in research areas including the food industry, blood bank safety and human and veterinary disease diagnosis.
New studies on the heat resistance of hamster-adapted scrapie agent: Threshold survival after ashing at 600°C suggests an inorganic template of replication
Prion Infected Meat-and-Bone Meal Is Still Infectious after Biodiesel Production
Detection of protease-resistant cervid prion protein in water from a CWD-endemic area
A Quantitative Assessment of the Amount of Prion Diverted to Category 1 Materials and Wastewater During Processing
Rapid assessment of bovine spongiform encephalopathy prion inactivation by heat treatment in yellow grease produced in the industrial manufacturing process of meat and bone meals
PPo4-4:
Survival and Limited Spread of TSE Infectivity after Burial
Discussion Classical scrapie is an environmentally transmissible disease because it has been reported in naïve, supposedly previously unexposed sheep placed in pastures formerly occupied by scrapie-infected sheep (4, 19, 20).
Although the vector for disease transmission is not known, soil is likely to be an important reservoir for prions (2) where – based on studies in rodents – prions can adhere to minerals as a biologically active form (21) and remain infectious for more than 2 years (22).
Similarly, chronic wasting disease (CWD) has re-occurred in mule deer housed in paddocks used by infected deer 2 years earlier, which was assumed to be through foraging and soil consumption (23).
Our study suggested that the risk of acquiring scrapie infection was greater through exposure to contaminated wooden, plastic, and metal surfaces via water or food troughs, fencing, and hurdles than through grazing.
Drinking from a water trough used by the scrapie flock was sufficient to cause infection in sheep in a clean building.
Exposure to fences and other objects used for rubbing also led to infection, which supported the hypothesis that skin may be a vector for disease transmission (9).
The risk of these objects to cause infection was further demonstrated when 87% of 23 sheep presented with PrPSc in lymphoid tissue after grazing on one of the paddocks, which contained metal hurdles, a metal lamb creep and a water trough in contact with the scrapie flock up to 8 weeks earlier, whereas no infection had been demonstrated previously in sheep grazing on this paddock, when equipped with new fencing and field furniture.
When the contaminated furniture and fencing were removed, the infection rate dropped significantly to 8% of 12 sheep, with soil of the paddock as the most likely source of infection caused by shedding of prions from the scrapie-infected sheep in this paddock up to a week earlier.
This study also indicated that the level of contamination of field furniture sufficient to cause infection was dependent on two factors: stage of incubation period and time of last use by scrapie-infected sheep.
Drinking from a water trough that had been used by scrapie sheep in the predominantly pre-clinical phase did not appear to cause infection, whereas infection was shown in sheep drinking from the water trough used by scrapie sheep in the later stage of the disease.
It is possible that contamination occurred through shedding of prions in saliva, which may have contaminated the surface of the water trough and subsequently the water when it was refilled.
Contamination appeared to be sufficient to cause infection only if the trough was in contact with sheep that included clinical cases.
Indeed, there is an increased risk of bodily fluid infectivity with disease progression in scrapie (24) and CWD (25) based on PrPSc detection by sPMCA.
Although ultraviolet light and heat under natural conditions do not inactivate prions (26), furniture in contact with the scrapie flock, which was assumed to be sufficiently contaminated to cause infection, did not act as vector for disease if not used for 18 months, which suggest that the weathering process alone was sufficient to inactivate prions.
PrPSc detection by sPMCA is increasingly used as a surrogate for infectivity measurements by bioassay in sheep or mice.
In this reported study, however, the levels of PrPSc present in the environment were below the limit of detection of the sPMCA method, yet were still sufficient to cause infection of in-contact animals.
In the present study, the outdoor objects were removed from the infected flock 8 weeks prior to sampling and were positive by sPMCA at very low levels (2 out of 37 reactions).
As this sPMCA assay also yielded 2 positive reactions out of 139 in samples from the scrapie-free farm, the sPMCA assay could not detect PrPSc on any of the objects above the background of the assay.
False positive reactions with sPMCA at a low frequency associated with de novo formation of infectious prions have been reported (27, 28).
This is in contrast to our previous study where we demonstrated that outdoor objects that had been in contact with the scrapie-infected flock up to 20 days prior to sampling harbored PrPSc that was detectable by sPMCA analysis [4 out of 15 reactions (12)] and was significantly more positive by the assay compared to analogous samples from the scrapie-free farm.
This discrepancy could be due to the use of a different sPMCA substrate between the studies that may alter the efficiency of amplification of the environmental PrPSc.
In addition, the present study had a longer timeframe between the objects being in contact with the infected flock and sampling, which may affect the levels of extractable PrPSc.
Alternatively, there may be potentially patchy contamination of this furniture with PrPSc, which may have been missed by swabbing.
The failure of sPMCA to detect CWD-associated PrP in saliva from clinically affected deer despite confirmation of infectivity in saliva-inoculated transgenic mice was associated with as yet unidentified inhibitors in saliva (29), and it is possible that the sensitivity of sPMCA is affected by other substances in the tested material.
In addition, sampling of amplifiable PrPSc and subsequent detection by sPMCA may be more difficult from furniture exposed to weather, which is supported by the observation that PrPSc was detected by sPMCA more frequently in indoor than outdoor furniture (12).
A recent experimental study has demonstrated that repeated cycles of drying and wetting of prion-contaminated soil, equivalent to what is expected under natural weathering conditions, could reduce PMCA amplification efficiency and extend the incubation period in hamsters inoculated with soil samples (30).
This seems to apply also to this study even though the reduction in infectivity was more dramatic in the sPMCA assays than in the sheep model.
Sheep were not kept until clinical end-point, which would have enabled us to compare incubation periods, but the lack of infection in sheep exposed to furniture that had not been in contact with scrapie sheep for a longer time period supports the hypothesis that prion degradation and subsequent loss of infectivity occurs even under natural conditions.
In conclusion, the results in the current study indicate that removal of furniture that had been in contact with scrapie-infected animals should be recommended, particularly since cleaning and decontamination may not effectively remove scrapie infectivity (31), even though infectivity declines considerably if the pasture and the field furniture have not been in contact with scrapie-infected sheep for several months. As sPMCA failed to detect PrPSc in furniture that was subjected to weathering, even though exposure led to infection in sheep, this method may not always be reliable in predicting the risk of scrapie infection through environmental contamination.
These results suggest that the VRQ/VRQ sheep model may be more sensitive than sPMCA for the detection of environmentally associated scrapie, and suggest that extremely low levels of scrapie contamination are able to cause infection in susceptible sheep genotypes.
Keywords: classical scrapie, prion, transmissible spongiform encephalopathy, sheep, field furniture, reservoir, serial protein misfolding cyclic amplification
Wednesday, December 16, 2015
*** Objects in contact with classical scrapie sheep act as a reservoir for scrapie transmission ***
WEDNESDAY, SEPTEMBER 19, 2018
CHRONIC WASTING DISEASE CWD TSE PRION Detection of a first case in Quebec Canada
THURSDAY, SEPTEMBER 13, 2018
NORWAY How many CWD-infected animals are out there?
WEDNESDAY, OCTOBER 03, 2018
Texas Reports 13 more cases of Chronic Wasting Disease CWD TSE Prion in Breeder Deer state total jumps to 130 Confirmed to date
TEXAS CONFIRMS 117TH CASE OF CWD TSE PRION
WEDNESDAY, AUGUST 22, 2018
TEXAS CWD TSE PRION 16 MORE CASES DETECTED TOTAL TO DATE 117 CONFIRMED NEW 14 BREEDERS 2 FREE RANGE
SUNDAY, AUGUST 19, 2018
Texas Deer Farms, Growing Freakish Antlers, and CWD TSE Prion aka mad deer disease
MONDAY, OCTOBER 01, 2018
Minnesota Deer tests positive for CWD in disease management zone
WEDNESDAY, OCTOBER 03, 2018
WISCONSIN CAVES TO GAME FARM INDUSTRY AGAIN WHILE STATE FALLS FURTHER INTO THE ABYSS OF MAD DEER DISEASE CWD TSE PRION
SATURDAY, SEPTEMBER 29, 2018
This Map Spells Trouble for the Future of Deer Hunting CWD TSE Prion Consumption, Exposure, and Zoonosis Potential
Michigan adds another CWD TSE Prion case, total at 63 to date
Total Deer Tested and Total Positives Cases 63
THURSDAY, OCTOBER 04, 2018
Michigan adds another CWD TSE Prion case, total at 63 to date
WEDNESDAY, SEPTEMBER 26, 2018
Michigan adds another CWD TSE Prion case, total at 62 to date
MONDAY, AUGUST 27, 2018
Michigan Adds Another CWD TSE Prion Case Total Increases To 61 to date
WEDNESDAY, SEPTEMBER 19, 2018
Michigan Department of Natural Resources Slowing the spread of CWD UPDATE Sept 19 2018
THURSDAY, JUNE 21, 2018
Michigan First case of chronic wasting disease suspected in Jackson County
THURSDAY, JUNE 07, 2018
Michigan DNR to present chronic wasting disease recommendations to Natural Resources Commission Singeltary submission
WEDNESDAY, MARCH 07, 2018
***> Michigan DNR CWD National Perspective: Captive Herd Certification Program - Dr. Tracy Nichols
***> CURRENT STATUS OF CWD IN CAPTIVE CERVID HERDS IN 16 STATES AS OF MAY 2017
43 ELK HERDS
37 WTD HERDS
1 RED DEER HERD
6 MIX SPECIES HERDS
85 CWD-POSITIVE CAPTIVE HERDS
snip...see
TUESDAY, MARCH 27, 2018
Hunters and citizens invited to collaborate on Michigan's chronic wasting disease response
FRIDAY, MARCH 30, 2018
Michigan Mecosta County man sentenced following DNR investigation Game ranch owner falsified information related to chronic wasting disease testing
what is Michigan feeding their cervid ???
2017 Section 21 C.F.R. 589.2000, Animal Proteins Prohibited in Ruminant Feed
Subject: MICHIGAN FDA PART 589 -- SUBSTANCES PROHIBITED FROM USE IN ANIMAL FOOD OR FEED VIOLATIONS OFFICIAL ACTION INDICATED OAI UPDATE BREACH APRIL 4, 2017
MICHIGAN FDA PART 589 -- SUBSTANCES PROHIBITED FROM USE IN ANIMAL FOOD OR FEEDVIOLATIONS OFFICIAL ACTION INDICATED OAI UPDATE BREACH APRIL 4, 2017
FDA BSE/Ruminant Feed Inspections Firms Inventory
11998 DET-DO MI 48846-847 OPR 4/4/2017 OAI
NAI = NO ACTION INDICATED
OAI = OFFICIAL ACTION INDICATED
VAI = VOLUNTARY ACTION INDICATED
RTS = REFERRED TO STATE
OAI (Official Action Indicated) when inspectors find significant objectionable conditions or practices and believe that regulatory sanctions are warranted to address the establishment’s lack of compliance with the regulation.. An example of an OAI classification would be findings of manufacturing procedures insufficient to ensure that ruminant feed is not contaminated with prohibited material. Inspectors will promptly re-inspect facilities classified OAI after regulatory sanctions have been applied to determine whether the corrective actions are adequate to address the objectionable conditions....end...TSS
WEDNESDAY, JULY 11, 2018
CONFIDENTIAL IN CONFIDENCE SPONGIFORM ENCEPHALOPATHY OF PIGS FDA EMERGENCY REQUEST FOR RULE CHANGE USA Section 21 C.F.R. 589.2000
SATURDAY, MARCH 03, 2018
Michigan-sportsman.com without warning bans terry singeltary sr. for posting peer review sound science on CWD TSE Prion after 16 years
LISTEN TO THIS NICE LITTLE CWD BLUES DIDDY BY TAMI ABOUT WISCONSIN CWD TSE PRION. WOW, ANNUAL UPDATES NOW, FROM HERE ON OUT, ABOUT CWD...200,000 CWD TESTS, WITH OVER 3500 CWD POSITIVE CASES, SEEING INCREASING TRENDS IN PREVALENCE AND DISTRIBUTION...CARCASS DISPOSAL SIGNIFICANT CHALLENGE...CWD SAMPLING EFFORTS GONE DONE, WHILE CWD POSITIVES HAVE GONE UP...ALSO, 40 SELF SERVING KIOSKS ACROSS STATE AND FREE HUNTER SERVICE CWD TESTING AND SICK DEER POLICY REPORTING AND TESTING ACROSS STATE!
TUESDAY, SEPTEMBER 25, 2018
Wisconsin CWD TSE PRION PLAN preferred option disposal in a landfill OR public land is acceptable to leave the carcass in the spot of the kill
stupid is, as stupid does, sometimes you can't fix stupid...tss
MONDAY, JUNE 25, 2018
Wisconsin DATCP Confirms CWD-Positive Elk in Sauk County Breeding Farm
MONDAY, JUNE 18, 2018
Wisconsin DATCP Confirms CWD-Positive Deer in Marinette County farm has been quarantined
WEDNESDAY, JUNE 13, 2018
Wisconsin DATCP NVSL confirmed 21 WTD from a deer farm Iowa County tested positive for chronic wasting disease (CWD)
SATURDAY, MARCH 03, 2018
WISCONSIN CHRONIC WASTING DISEASE TSE Prion DNR Study Finds CWD-Infected Deer Die At 3 Times Rate Of Healthy Animals
FRIDAY, FEBRUARY 16, 2018
Wisconsin Deer from Now-Quarantined PA Lancaster County Farm Tests Positive for Chronic Wasting Disease CWD TSE Prion
FRIDAY, JANUARY 26, 2018
WISCONSIN REPORTS 588 CWD TSE PRION POSITIVE CASES FOR 2017 WITH 4170 CASES CONFIRMED TO DATE
USA MAD DEER ROUNDUP
Feb. 16, 2018
Durkin: Stop private deer industry from trucking CWD across state
Patrick Durkin, For USA TODAY NETWORK-Wisconsin Published 10:13 a.m. CT Feb. 16, 2018
A Waupaca County captive-deer shooting preserve that discovered its first two cases of chronic wasting disease in October found 10 more CWD cases last fall, with 11 of the deer coming from a breeding facility in Iowa County — Wisconsin’s most infected county.
Hunt’s End Deer Ranch near Ogdensburg is one of 376 fenced deer farms in Wisconsin, according to the Department of Agriculture, Trade and Consumer Protection. Hunt’s End bought the diseased deer from Windy Ridge Whitetails, a 15-acre, 110-deer breeding facility south of Mineral Point in Iowa County. Of Wisconsin’s 4,175 CWD cases in wild deer, 2,261 (54 percent) are in Iowa County.
Since CWD’s discovery in three wild deer shot during the November 2001 gun season, CWD has been detected on 18 Wisconsin deer farms, of which 11 were “depopulated.” DATCP has identified 242 CWD cases in captive facilities the past 16 years.
The state’s worst site remains the former Buckhorn Flats Game Farm near Almond in Portage County, where 80 deer tested positive for this always-fatal disease from 2002 to 2006. When the U.S. Department of Agriculture shot out the 70-acre pen in January 2006, 60 of the remaining 76 deer carried CWD, a nearly 80 percent infection rate.
The Department of Natural Resources bought the heavily contaminated site for $465,000 in 2011 and has kept it fenced and deer-free since.
The last time DATCP exterminated a captive herd was November 2015, when it killed 228 deer at Fairchild Whitetails, a 10-acre breeding facility in Eau Claire County, and paid its owner, Richard Vojtik, $298,770 in compensation. Tests revealed 34 of those deer carried CWD (15 percent), but two bucks had escaped earlier. Those bucks roamed five months before being shot and tested. They, too, had CWD.
Both operations were outside the endemic CWD region in southern Wisconsin; Buckhorn Flats by about 60 miles and Fairchild Whitetails by about 120. Wisconsin’s four most active CWD outbreaks on deer farms are north of U.S. 10, and farther away from the endemic region — basically the DNR’s Southern Farmlands district — which had 584 CWD cases 2017-18 and 4,148 since 2001.
Those businesses are:
• Wilderness Whitetails, near Eland in Marathon County: 68 CWD cases, including 43 in 2017-18. DATCP first reported CWD there in December 2013 in a 5-year-old buck shot by a facility client. The operation also found three cases in 2014, nine in 2015 and 12 in 2016.
The preserve held about 310 deer in its 351-acre pen last summer. Since beginning tests in 2002, the facility tested 373 deer before finding its first case 11 years later.
• Hunt’s End, Waupaca County: 12 cases, all in 2017-18. The owners, Dusty and Mandy Reid, didn’t detect CWD on the 84-acre shooting facility until two 4-year-old bucks tested positive last fall. DATCP announced those cases Oct. 20, and disclosed 10 additional cases in response to my open-records request in January.
Both Oct. 20 bucks originated from Windy Ridge Whitetails. Nine other bucks from Windy Ridge, owned by Steven and Marsh Bertram, tested positive for CWD after being shot by Hunt’s End clients.
Now DATCP records covering the past five years showed Hunt’s End acquired 31 deer from Windy Ridge, which also sent a combined 67 whitetails to nine other Wisconsin deer farms during that period.
Paul McGraw, DATCP’s state veterinarian and administrator in animal health, quarantined three Hunt’s End properties Oct. 20, but let its owners, continue selling hunts because “properly handled dead animals leaving the premises do not pose a disease risk.”
McGraw also quarantined Windy Ridge, but the specifications let the business move more deer to the Waupaca shooting facility. It made two more shipments to Hunt’s End, the last occurring Nov. 13.
• Apple Creek Whitetails, Oconto County: 11 cases. Since discovering CWD in September 2016 in an 18-month-old doe killed inside the facility near Gillett, DATCP has identified 10 more cases, including three in 2017-18. The preserve held about 1,850 deer on 1,363 acres, and tested 466 in 2016. After first testing for CWD in 2009, the business processed 1,192 deer before finding its first case 18 months ago.
• Three Lakes Trophy Ranch, Oneida County: Nine cases. Since discovering CWD in December 2015 in a 3-year-old buck at Three Lakes, DATCP has identified eight more cases, including two in 2017-18. The preserve held about 545 whitetails on 570 acres.
Although the Hunt’s End outbreak traces to Iowa County deer, Windy Ridge Whitetails sent even more deer, 42, to Vojtik’s American Adventures Ranch near Fairchild with no documented problems. DATCP reports no CWD cases there, and Vojtik, who also owned the 10-acre Fairchild Whitetails breeding facility, said he hasn’t bought Windy Ridge deer the past two years.
Vojtik said Wednesday that he and his clients shoot out his enclosure’s herd of about 200 deer each year to reduce CWD risks. And because he’s not in DATCP’s herd-status program, he must only test 50 percent of deer dying there.
Meanwhile, Wilderness Whitetails tests all of its dead deer. It leads the state with 68 CWD cases, even though it has maintained a “closed herd” since opening its Eland facility in 2004, said its owner, Greg Flees, when reached Wednesday. Flees said all deer in the 351-acre facility were born there or came from his family’s Portage County breeding pen, which began in the 1970s and has never had CWD.
Flees said the jump from 12 CWD cases in 2016 to 43 in 2017 is no mystery or surprise. “We shot more deer to lower our densities, so we found more CWD,” he said. He thinks CWD was in the facility’s soils when they enclosed it with an 8-foot-high fence 14 years ago, or it arrived in alfalfa bales brought in for feed.
Perhaps the bigger mystery is why DATCP allows any deer from Iowa County to be shipped anywhere. Windy Ridge Whitetails is one of eight captive-deer facilities in CWD-infected counties — Sauk, Dane, Iowa, Rock, Walworth and Richland — enrolled in DATCP’s herd-status program, which allows deer transfers if facilities follow specified guidelines.
That won’t change soon, either. In a letter Jan. 30 responding to my open records request, Paul Dedinsky, DATCP’s chief legal counsel, wrote, “The Department is not proposing any rule changes to prohibit movement from CWD endemic areas.”
No doubt Wisconsin’s wild deer provide a vast, mostly undocumented pool for spreading CWD, but sick deer can only carry disease as far as they walk. With DATCP’s approval, privately owned deer could spread CWD wherever they’re trucked.
Patrick Durkin is a freelance writer who covers outdoors for USA TODAY NETWORK-Wisconsin.. Email him at patrickdurkin56@gmail.com.
FRIDAY, FEBRUARY 16, 2018
Wisconsin Stop private deer industry from trucking CWD across state
Tuesday, December 20, 2011
CHRONIC WASTING DISEASE CWD WISCONSIN Almond Deer (Buckhorn Flats) Farm Update DECEMBER 2011
The CWD infection rate was nearly 80%, the highest ever in a North American captive herd. RECOMMENDATION: That the Board approve the purchase of 80 acres of land for $465,000 for the Statewide Wildlife Habitat Program in Portage County and approve the restrictions on public use of the site.
SUMMARY:
***>captive deer farmers breeders entitlement program, i.e. indemnity program, why?
how many states have $465,000., and can quarantine and purchase there from, each cwd said infected farm, but how many states can afford this for all the cwd infected cervid game ranch type farms, and why do tax payers have to pay for it ???
MONDAY, MARCH 26, 2018
Wisconsin Rep. Milroy Wants More Action to Combat CWD TSE Prion aka Mad Deer Disease
THURSDAY, SEPTEMBER 13, 2018
Two Louisiana Residents Plead Guilty to Smuggling Live White-Tailed Deer into Mississippi that came from a herd of captive white-tailed deer in Pennsylvania that tested positive for Chronic Wasting Disease CWD
THURSDAY, OCTOBER 04, 2018
Colorado Parks and Wildlife seeks input on chronic wasting disease plan
Deer Antler Velvet and the CWD TSE Prion
Volume 15, Number 5—May 2009
Research
Chronic Wasting Disease Prions in Elk Antler Velvet
Rachel C. Angers1, Tanya S. Seward, Dana Napier, Michael Green, Edward Hoover, Terry Spraker, Katherine O’Rourke, Aru Balachandran, and Glenn C. TellingComments to Author Author affiliations: University of Kentucky Medical Center, Lexington, Kentucky, USA (R.C. Angers, T.S. Seward, D. Napier, M. Green, G.C. Telling); Colorado State University, Fort Collins, Colorado, USA (E. Hoover, T. Spraker); US Department of Agriculture, Pullman, Washington, USA (K. O’Rourke); Canadian Food Inspection Agency, Ottawa, Ontario, Canada (A. Balachandran); 1Current affiliation: Medical Research Council Laboratory of Molecular Biology, Cambridge, UK.
Abstract
Chronic wasting disease (CWD) is a contagious, fatal prion disease of deer and elk that continues to emerge in new locations. To explore the means by which prions are transmitted with high efficiency among cervids, we examined prion infectivity in the apical skin layer covering the growing antler (antler velvet) by using CWD-susceptible transgenic mice and protein misfolding cyclic amplification. Our finding of prions in antler velvet of CWD-affected elk suggests that this tissue may play a role in disease transmission among cervids. Humans who consume antler velvet as a nutritional supplement are at risk for exposure to prions. The fact that CWD prion incubation times in transgenic mice expressing elk prion protein are consistently more rapid raises the possibility that residue 226, the sole primary structural difference between deer and elk prion protein, may be a major determinant of CWD pathogenesis.
Sunday, September 16, 2018
Mother to Offspring Transmission of TSE PRION DISEASE and risk factors there from
TUESDAY, AUGUST 07, 2018
Passage of scrapie to deer results in a new phenotype upon return passage to sheep
THURSDAY, SEPTEMBER 27, 2018
Estimating the impact on food and edible materials of changing scrapie control measures: The scrapie control model
MONDAY, OCTOBER 01, 2018
Update on Classical and Atypical Scrapie in Sheep and Goats: Review 2018
TUESDAY, SEPTEMBER 4, 2018
USA CJD, BSE, SCRAPIE, CWD, TSE PRION END OF YEAR REPORTS September 4, 2018
ONE more thing, please remember, the label does not have to say ''deer ration'' for cervid to be pumped up with. you can get the same ''high protein'' from many sources of high protein feed for animals other than cattle, and feed them to cervid...
Saturday, August 29, 2009
FOIA REQUEST FEED RECALL 2009 Product may have contained prohibited materials Bulk Whole Barley, Recall # V-256-2009
Friday, September 4, 2009
FOIA REQUEST ON FEED RECALL PRODUCT 429,128 lbs. feed for ruminant animals may have been contaminated with prohibited material Recall # V-258-2009
WEDNESDAY, JULY 11, 2018
CONFIDENTIAL IN CONFIDENCE SPONGIFORM ENCEPHALOPATHY OF PIGS FDA EMERGENCY REQUEST FOR RULE CHANGE USA Section 21 C.F.R. 589.2000
TUESDAY, JULY 10, 2018
CONFIDENTIAL IN CONFIDENCE SPONGIFORM ENCEPHALOPATHY OF PIGS
*** ''but feeding of other ruminant protein, including scrapie-infected sheep, can continue to pigs.''
CONFIDENTIAL SPONGIFORM ENCEPHALOPATHY OF PIGS
SUNDAY, SEPTEMBER 23, 2018
Low-volume goat milk transmission of classical scrapie to lambs and goat kids
WEDNESDAY, SEPTEMBER 19, 2018
CHRONIC WASTING DISEASE CWD TSE PRION Detection of a first case in Quebec Canada
TUESDAY, JULY 03, 2018
Chronic Wasting Disease CWD TSE Prion Global Report Update, USA, CANADA, KOREA, NORWAY, FINLAND, Game Farms and Fake news
SUNDAY, APRIL 8, 2018
Transmissible Spongiform Encephalopathy TSE Prion Disease Global Pandemic Urgent Update April 9, 2018
USDA ONLY TESTING 20k HEAD OF CATTLE A YEAR FOR MAD COW DISEASE ...LOL!
WEDNESDAY, AUGUST 29, 2018
USDA Announces Atypical Bovine Spongiform Encephalopathy Detection USDA 08/29/2018 10:00 AM EDT
WEDNESDAY, AUGUST 29, 2018
USDA DROPS MAD COW TESTING FROM 40K A YEAR TO JUST 20K A YEAR, IMPOSSIBLE TO FIND BSE, BUT THEY DID, IN FLORIDA!
''Atypical BSE is different, and it generally occurs in older cattle, usually 8 years of age or greater. It seems to arise rarely and spontaneously in all cattle populations.''
FALSE!
''The primary source of infection for classical BSE is feed contaminated with the infectious prion agent, such as meat-and-bone meal containing protein derived from rendered infected cattle. Regulations from the Food and Drug Administration (FDA) have prohibited the inclusion of mammalian protein in feed for cattle and other ruminants since 1997 and have also prohibited high risk tissue materials in all animal feed since 2009.''
FALSE!
LET'S REVIEW RECENT AND PAST SCIENCE THAT SHOWS THE ABOVE TWO STATEMENTS ARE FAR FROM TRUE;
PRION 2018 CONFERENCE
P98 The agent of H-type bovine spongiform encephalopathy associated with E211K prion protein polymorphism transmits after oronasal challenge
Greenlee JJ (1), Moore SJ (1), and West Greenlee MH (2)
(1) United States Department of Agriculture, Agricultural Research Service, National Animal Disease Center, Virus and Prion Research Unit, Ames, IA, United States (2) Department of Biomedical Sciences, Iowa State University College of Veterinary Medicine, Ames, IA, United States.
In 2006, a case of H-type bovine spongiform encephalopathy (BSE) was reported in a cow with a previously unreported prion protein polymorphism (E211K).
The E211K polymorphism is heritable and homologous to the E200K mutation in humans that is the most frequent PRNP mutation associated with familial Creutzfeldt-Jakob disease.
Although the prevalence of the E211K polymorphism is low, cattle carrying the K211 allele develop H-type BSE with a rapid onset after experimental inoculation by the intracranial route.
The purpose of this study was to investigate whether the agents of H-type BSE or H-type BSE associated with the E211K polymorphism transmit to wild type cattle or cattle with the K211 allele after oronasal exposure.
Wild type (EE211) or heterozygous (EK211) cattle were oronasally inoculated with either H-type BSE from the 2004 US Htype BSE case (n=3) or from the 2006 US H-type case associated with the E211K polymorphism (n=4) using 10% w/v brain homogenates.
Cattle were observed daily throughout the course of the experiment for the development of clinical signs.
At approximately 50 months post-inoculation, one steer (EK211 inoculated with E211K associated H-BSE) developed clinical signs including inattentiveness, loss of body condition, weakness, ataxia, and muscle fasciculations and was euthanized.
Enzyme immunoassay confirmed that abundant misfolded protein was present in the brainstem, and immunohistochemistry demonstrated PrPSc throughout the brain.
Western blot analysis of brain tissue from the clinically affected steer was consistent with the E211K H-type BSE inoculum.
With the experiment currently at 55 months post-inoculation, no other cattle in this study have developed clinical signs suggestive of prion disease.
This study demonstrates that the H-type BSE agent is transmissible by the oronasal route.
These results reinforce the need for ongoing surveillance for classical and atypical BSE to minimize the risk of potentially infectious tissues entering the animal or human food chains.
=====
O10 Zoonotic potential of atypical BSE prions: a systematic evaluation
Marín-Moreno A (1), Espinosa JC (1), Douet JY (2), Aguilar-Calvo P (1), Píquer J (1), Lorenzo P (1), Lacroux C (2), Huor A (2), Lugan S (2), Tillier C (2), Andreoletti O (2) and Juan María Torres (1)
(1) Centro de Investigación en Sanidad Animal, CISA-INIA, Carretera Algete-El Casar s/n, Valdeolmos, 28130 Madrid, Spain.(2) UMR INRA ENVT 1225, Interactions Hôtes Agents Pathogènes, Ecole Nationale Vétérinaire de Toulouse, France.
Bovine Spongiform Encephalopathy (BSE) is the only zoonotic prion recognized to date. The transmission of BSE to humans caused the emergence of variant Creutzfeldt-Jakob disease (vCJD). In 2004 two new atypical prion agents were identified in cattle: H- and L- BSE prion strains.
The zoonotic potential of atypical BSE prions was assessed by inoculating three different isolates of cattle H- and L-BSE in transgenic mouse lines that overexpress the human PrP covering the three different genotypes of the aminoacid 129 (TgMet129, TgMet/Val129 and TgVal129). This polymorphism is known to be a key element involved in human resistance/susceptibility to BSE. In addition, TgMet129 and TgVal129 were challenged with one H- and L-BSE isolates adapted to sheep PrP expressing hosts to assess if intermediate passage in sheep could modify the capacity of these prions to cross the human species barrier.
Our results confirm that L-BSE transmits to TgMet129 even better than epidemic BSE. However, atypical L-BSE agent was unable to infect TgVal129 or TgMet/Val129 mice, even after passage in TgMet129. No transmission was observed with H-BSE in any mice model inoculated, irrespectively of the 129 polymorphism. After passage in sheep PrP expressing host, the properties of both H and LBSE including their capacity to cross the human species barrier were dramatically affected, emerging prion strains features that resemble those of sporadic Creutzfeldt-Jakob disease (sCJD).
To date, this is the more extensive and complete analysis of the zoonotic potential of atypical BSE prions. These results advise not to ignore the zoonotic potential of these agents.
=====
P77 In vitro approach to estimate the human transmission risk of prions
Iwamaru Y (1) Imamura M (2) Matsuura Y (1) Kohtaro Miyazawa (1) Takashi Yokoyama (3)
(1 ) National Institute of Animal Health, Prion Disease Unit, Ibaraki, Japan (2) University of Miyazaki, Division of Microbiology, Miyazaki, Japan (3) National Institute of Animal Health, Department of Planning and General Administration, Ibaraki, Japan.
Prion diseases are fatal neurodegenerative disorders in humans and animals. The key event in the pathogenesis of these disease is the conversion of host-encoded normal cellular prion protein (PrPC) into its pathogenic isoform (PrPSc) and its accumulation in the central nervous system. One of the characteristics of prion is the species barrier that limits the transmission between different species. Currently, bioassays using transgenic mice (Tg) overexpressing PrP of different species have become valuable tools for assessing cross species transmissibility of prions.
The recent reports describing the emergence of novel bovine spongiform encephalopathy (BSE) from H-BSE and the transmission of chronic wasting disease to swine have generated concerns of human infections of newly identified prions. Although Tg expressing human PrP have been used to model human susceptibility to animal prions, these experiments are costly and time-consuming. In addition, the results of bioassays are influenced by the lines of transgenic mice used and the lifespan of the challenged animals. These factors are needed to be taken into account when assessing the human risk of prions.
In attempt to develop the more time- and cost-saving method for assessment of the human transmission risk of prions, we performed experiments using protein misfolding cyclic amplification (PMCA) technique to investigate whether PMCA can be compatible with bioassay. Using brain homogenates of Tg expressing bovine PrP as the PrP substrate, we optimized the versatile PMCA condition that could amplify PrPSc from cattle affected with C-, H- or L-BSE. We measured the 50% PMCA seeding activity dose and the 50% lethal dose in 1 g equivalent of C-, H- or L-BSE cattle brain tissue by using PMCA or bioassay, respectively, and assessed the correlations between these doses.
=====
P98 The agent of H-type bovine spongiform encephalopathy associated with E211K prion protein polymorphism transmits after oronasal challenge
Greenlee JJ (1), Moore SJ (1), and West Greenlee MH (2) (1) United States Department of Agriculture, Agricultural Research Service, National Animal Disease Center, Virus and Prion Research Unit, Ames, IA, United States (2) Department of Biomedical Sciences, Iowa State University College of Veterinary Medicine, Ames, IA, United States.
reading up on this study from Prion 2018 Conference, very important findings ;
***> This study demonstrates that the H-type BSE agent is transmissible by the oronasal route.
***> These results reinforce the need for ongoing surveillance for classical and atypical BSE to minimize the risk of potentially infectious tissues entering the animal or human food chains.
PRION 2018 CONFERENCE ABSTRACT
WEDNESDAY, AUGUST 15, 2018
The agent of H-type bovine spongiform encephalopathy associated with E211K prion protein polymorphism transmits after oronasal challenge
Evidence That Transmissible Mink Encephalopathy Results from Feeding Infected Cattle
Over the next 8-10 weeks, approximately 40% of all the adult mink on the farm died from TME.
snip...
The rancher was a ''dead stock'' feeder using mostly (>95%) downer or dead dairy cattle...
http://web.archive.org/web/20090506024922/http://www.bseinquiry.gov.uk/files/yb/1987/06/10004001.pdf
In Confidence - Perceptions of unconventional slow virus diseases of animals in the USA - APRIL-MAY 1989 - G A H Wells
3. Prof. A. Robertson gave a brief account of BSE. The US approach was to accord it a very low profile indeed. Dr. A Thiermann showed the picture in the ''Independent'' with cattle being incinerated and thought this was a fanatical incident to be avoided in the US at all costs. ...
”The occurrence of CWD must be viewed against the contest of the locations in which it occurred. It was an incidental and unwelcome complication of the respective wildlife research programmes. Despite it’s subsequent recognition as a new disease of cervids, therefore justifying direct investigation, no specific research funding was forthcoming. The USDA veiwed it as a wildlife problem and consequently not their province!” ...page 26.
*** Spraker suggested an interesting explanation for the occurrence of CWD. The deer pens at the Foot Hills Campus were built some 30-40 years ago by a Dr. Bob Davis. At or abut that time, allegedly, some scrapie work was conducted at this site. When deer were introduced to the pens they occupied ground that had previously been occupied by sheep.
***>2018<***
TUESDAY, AUGUST 7, 2018
Unexpected prion phenotypes in experimentally transfused animals: predictive models for humans?
***> NEW TRANSMISSIBLE SPONGIFORM ENCEPHALOPATHY TSE PRION DISEASE (MAD CAMEL DISEASE) IN A NEW SPECIES <***
NEW OUTBREAK OF TRANSMISSIBLE SPONGIFORM ENCEPHALOPATHY TSE PRION DISEASE IN A NEW SPECIES
Subject: Prion Disease in Dromedary Camels, Algeria
Our identification of this prion disease in a geographically widespread livestock species requires urgent enforcement of surveillance and assessment of the potential risks to human and animal health.
Wednesday, May 30, 2018
Dromedary camels in northern Africa have a neurodegenerative prion disease that may have originated decades ago
***> IMPORTS AND EXPORTS <***
SEE MASSIVE AMOUNTS OF BANNED ANIMAL PROTEIN AKA MAD COW FEED IN COMMERCE USA DECADES AFTER POST BAN
WEDNESDAY, SEPTEMBER 26, 2018
***> JAVMA In Short Update USDA announces detection of atypical BSE
TUESDAY, SEPTEMBER 4, 2018
USA CJD, BSE, SCRAPIE, CWD, TSE PRION END OF YEAR REPORTS September 4, 2018
WEDNESDAY, SEPTEMBER 5, 2018
APHIS Concurrence With OIE Risk Designations for Bovine Spongiform Encephalopathy [Docket No. APHIS-2018-0012]
HUMAN MAD COW DISEASE nvCJD TEXAS CASE NOT LINKED TO EUROPEAN TRAVEL CDC
Sunday, November 23, 2014
Confirmed Variant Creutzfeldt-Jakob Disease (variant CJD) Case in Texas
Updated: October 7, 2014
CDC and the Texas Department of State Health Services (DSHS) have completed the investigation of the recently reported fourth vCJD case in the United States. It confirmed that the case was in a US citizen born outside the Americas and indicated that the patient's exposure to the BSE/vCJD agent most likely occurred before he moved to the United States; the patient had resided in Kuwait, Russia and Lebanon. The completed investigation did not support the patient's having had extended travel to European countries, including the United Kingdom, or travel to Saudi Arabia. The specific overseas country where this patient’s infection occurred is less clear largely because the investigation did not definitely link him to a country where other known vCJD cases likely had been infected.
https://www.cdc.gov/ncidod/dvrd/vcjd/other/confirmed-case-in-texas.htm
https://vcjd.blogspot.com/2014/11/confirmed-variant-creutzfeldt-jakob.html
WEDNESDAY, SEPTEMBER 05, 2018
Edmonton woman one of the youngest diagnosed with CJD at 35 years old and pregnant
WEDNESDAY, SEPTEMBER 26, 2018
A new variant of Creutzfeldt-Jakob disease in the UK 1995 revisited 2018 a review of science
CBC news
*** USA sporadic CJD MAD COW DISEASE HAS HUGE PROBLEM Video
*** sporadic CJD linked to mad cow disease
*** you can see video here and interview with Jeff's Mom, and scientist telling you to test everything and potential risk factors for humans ***
1997-11-10: Panorama - The british disease
*** Human Mad Cow Video
TUESDAY, JULY 31, 2018
USA CJD TSE Tables of Cases Examined National Prion Disease Pathology Surveillance Center Cases Examined May 1, 2018
http://prionunitusaupdate.blogspot.com/2018/07/usa-cjd-tse-tables-of-cases-examined.html
THURSDAY, OCTOBER 04, 2018
National Prion Disease Pathology Surveillance Center Cases Examined¹ (September 18, 2018)
*** Conclusions. Preliminary results from transmission studies in bank voles strongly support the notion that VPSPr is a transmissible prion disease. Interestingly, VPSPr undergoes divergent evolution in the two genetic lines of voles, with sCJD-like features in BvM109 and GSS-like properties in BvI109. The discovery of previously unrecognized prion diseases in both humans and animals (i.e., Nor98 in small ruminants) demonstrates that the range of prion diseases might be wider than expected and raises crucial questions about the epidemiology and strain properties of these new forms. We are investigating this latter issue by molecular and biological comparison of VPSPr, GSS and Nor98.
Romolo Nonno,1 Michele Di Bari,1 Laura Pirisinu,1 Claudia D’Agostino,1 Stefano Marcon,1 Geraldina Riccardi,1 Gabriele Vaccari,1 Piero Parchi,2 Wenquan Zou,3 Pierluigi Gambetti,3 Umberto Agrimi1 1 Istituto Superiore di Sanità; Rome, Italy; 2 Dipartimento di Scienze Neurologiche, Università di Bologna; Bologna, Italy; 3 Case Western Reserve University; Cleveland, OH USA
Background. Variably protease-sensitive prionopathy (VPSPr) is a recently described “sporadic”neurodegenerative disease involving prion protein aggregation, which has clinical similarities with non-Alzheimer dementias, such as fronto-temporal dementia. Currently, 30 cases of VPSPr have been reported in Europe and USA, of which 19 cases were homozygous for valine at codon 129 of the prion protein (VV), 8 were MV and 3 were MM. A distinctive feature of VPSPr is the electrophoretic pattern of PrPSc after digestion with proteinase K (PK). After PK-treatment, PrP from VPSPr forms a ladder-like electrophoretic pattern similar to that described in GSS cases. The clinical and pathological features of VPSPr raised the question of the correct classification of VPSPr among prion diseases or other forms of neurodegenerative disorders. Here we report preliminary data on the transmissibility and pathological features of VPSPr cases in bank voles.
Materials and Methods. Seven VPSPr cases were inoculated in two genetic lines of bank voles, carrying either methionine or isoleucine at codon 109 of the prion protein (named BvM109 and BvI109, respectively). Among the VPSPr cases selected, 2 were VV at PrP codon 129, 3 were MV and 2 were MM. Clinical diagnosis in voles was confirmed by brain pathological assessment and western blot for PK-resistant PrPSc (PrPres) with mAbs SAF32, SAF84, 12B2 and 9A2.
Results. To date, 2 VPSPr cases (1 MV and 1 MM) gave positive transmission in BvM109. Overall, 3 voles were positive with survival time between 290 and 588 d post inoculation (d.p.i.). All positive voles accumulated PrPres in the form of the typical PrP27–30, which was indistinguishable to that previously observed in BvM109 inoculated with sCJDMM1 cases. In BvI109, 3 VPSPr cases (2 VV and 1 MM) showed positive transmission until now. Overall, 5 voles were positive with survival time between 281 and 596 d.p.i.. In contrast to what observed in BvM109, all BvI109 showed a GSS-like PrPSc electrophoretic pattern, characterized by low molecular weight PrPres. These PrPres fragments were positive with mAb 9A2 and 12B2, while being negative with SAF32 and SAF84, suggesting that they are cleaved at both the C-terminus and the N-terminus. Second passages are in progress from these first successful transmissions.
Conclusions. Preliminary results from transmission studies in bank voles strongly support the notion that VPSPr is a transmissible prion disease. Interestingly, VPSPr undergoes divergent evolution in the two genetic lines of voles, with sCJD-like features in BvM109 and GSS-like properties in BvI109. The discovery of previously unrecognized prion diseases in both humans and animals (i.e., Nor98 in small ruminants) demonstrates that the range of prion diseases might be wider than expected and raises crucial questions about the epidemiology and strain properties of these new forms. We are investigating this latter issue by molecular and biological comparison of VPSPr, GSS and Nor98.
BMJ Case Reports 2017; doi:10.1136/bcr-2017-220907
Rare disease
CASE REPORT
Gerstmann-Sträussler-Scheinker disease with atypical presentation
Sarah E Keuss1, James W Ironside2, Jonathan O’Riordan1
+ Author Affiliations
1Department of Neurology, Ninewells Hospital, Dundee, Tayside, UK
2Department of Clinical Brain Sciences, National Creutzfeldt-Jakob Disease Research and Surveillance Unit, Edinburgh, UK
Correspondence to Dr Jonathan O’Riordan, joriordan@nhs.net
Accepted 6 September 2017
Published 1 November 2017
Summary
We describe a 37-year-old woman who presented with progressive deafness, visual loss and ataxia. She latterly developed neuropsychiatric problems, including cognitive impairment, paranoid delusions and episodes of altered consciousness. She was found to be heterozygous for the Q212P mutation in the prion protein gene. She died over a decade after initial presentation and a diagnosis of prion disease was confirmed at postmortem.
Gerstmann-Sträussler-Scheinker disease subtypes efficiently transmit in bank voles as genuine prion diseases
Published online: 04 February 2016
Tuesday, November 29, 2016
Transmissibility of Gerstmann–Sträussler–Scheinker syndrome in rodent models: new insights into the molecular underpinnings of prion infectivity
2015 PRION CONFERENCE
*** RE-P.164: Blood transmission of prion infectivity in the squirrel monkey: The Baxter study
***suggest that blood donations from cases of GSS (and perhaps other familial forms of TSE) carry more risk than from vCJD cases, and that little or no risk is associated with sCJD. ***
P.164: Blood transmission of prion infectivity in the squirrel monkey: The Baxter study
Paul Brown1, Diane Ritchie2, James Ironside2, Christian Abee3, Thomas Kreil4, and Susan Gibson5 1NIH (retired); Bethesda, MD USA; 2University of Edinburgh; Edinburgh, UK; 3University of Texas; Bastrop, TX USA; 4Baxter Bioscience; Vienna, Austria; 5University of South Alabama; Mobile, AL USA
Five vCJD disease transmissions and an estimated 1 in 2000 ‘silent’ infections in UK residents emphasize the continued need for information about disease risk in humans. A large study of blood component infectivity in a non-human primate model has now been completed and analyzed. Among 1 GSS, 4 sCJD, and 3 vCJD cases, only GSS leukocytes transmitted disease within a 5–6 year surveillance period. A transmission study in recipients of multiple whole blood transfusions during the incubation and clinical stages of sCJD and vCJD in ic-infected donor animals was uniformly negative. These results, together with other laboratory studies in rodents and nonhuman primates and epidemiological observations in humans, suggest that blood donations from cases of GSS (and perhaps other familial forms of TSE) carry more risk than from vCJD cases, and that little or no risk is associated with sCJD. The issue of decades-long incubation periods in ‘silent’ vCJD carriers remains open.
ran across an old paper from 1984 ;
***The occurrence of contact cases raises the possibility that transmission in families may be effected by an unusually virulent strain of the agent. ***
snip...see full text ;
Variably protease-sensitive prionopathy (VPSPr), a recently identified and seemingly sporadic human prion disease, is distinct from Creutzfeldt-Jakob disease (CJD) but shares features of Gerstmann-Sträussler-Scheinker disease (GSS). However, contrary to exclusively inherited GSS, no prion protein (PrP) gene variations have been detected in VPSPr, suggesting that VPSPr might be the long-sought sporadic form of GSS. snip...
In conclusion, we propose that VPSPr is transmissible and, therefore, is an authentic prion disease. However, transmissibility cannot be sustained through serial passages presumably because human PrPC (or the mouse brain environment) cannot efficiently convert and propagate the VPSPr PrPSc species. If this is the case, uncovering the properties of human PrP that are required to replicate more efficiently the prion strains associated with VPSPr may help clarify the PrPSc mode of formation in this intriguing disease.
WEDNESDAY, NOVEMBER 09, 2011
Case report Sporadic fatal insomnia in a young woman: A diagnostic challenge: Case Report TEXAS
HOW TO TURN A POTENTIAL MAD COW VICTIM IN THE USA, INTO A HAPPENSTANCE OF BAD LUCK, A SPONTANEOUS MUTATION FROM NOTHING.
OR WAS IT $$$
FRIDAY, NOVEMBER 3, 2017
GSS Gerstmann-Sträussler-Scheinker disease with atypical presentation
WEDNESDAY, SEPTEMBER 26, 2018
A new variant of Creutzfeldt-Jakob disease in the UK 1995 revisited 2018 a review of science
Friday, January 10, 2014
***> vpspr, sgss, sffi, TSE, an iatrogenic by-product of gss, ffi, familial type prion disease, what it ???
*** ALL iatrogenic cjd is, is sporadic cjd, until the iatrogenic event is discovered, traced back, documented in the Academic domain, and then put into the public domain and documented as an iatrogenic CJD event. that’s why 85%+ of all human TSE prion disease is still sporadic CJD. problem solved $$$
PLEASE REMEMBER, IN 55 YEARS AND OLDER, THE RATE OF DOCUMENTED CJD JUMPS TO ONE IN 9,000.
Diagnosis and Reporting of Creutzfeldt-Jakob Disease
Singeltary, Sr et al. JAMA.2001; 285: 733-734. Vol. 285 No. 6, February 14, 2001 JAMA Diagnosis and Reporting of Creutzfeldt-Jakob Disease
To the Editor:
In their Research Letter, Dr Gibbons and colleagues1 reported that the annual US death rate due to Creutzfeldt-Jakob disease (CJD) has been stable since 1985. These estimates, however, are based only on reported cases, and do not include misdiagnosed or preclinical cases. It seems to me that misdiagnosis alone would drastically change these figures. An unknown number of persons with a diagnosis of Alzheimer disease in fact may have CJD, although only a small number of these patients receive the postmortem examination necessary to make this diagnosis. Furthermore, only a few states have made CJD reportable. Human and animal transmissible spongiform encephalopathies should be reportable nationwide and internationally.
Terry S. Singeltary, Sr Bacliff, Tex
1. Gibbons RV, Holman RC, Belay ED, Schonberger LB. Creutzfeldt-Jakob disease in the United States: 1979-1998. JAMA. 2000;284:2322-2323.
Tracking spongiform encephalopathies in North America
Xavier Bosch
Published: August 2003
Summary;
“My name is Terry S Singeltary Sr, and I live in Bacliff, Texas. I lost my mom to hvCJD (Heidenhain variant CJD) and have been searching for answers ever since. What I have found is that we have not been told the truth. CWD in deer and elk is a small portion of a much bigger problem.”
49-year-old Singeltary is one of a number of people who have remained largely unsatisfied after being told that a close relative died from a rapidly progressive dementia compatible with spontaneous Creutzfeldt-Jakob disease (CJD). So he decided to gather hundreds of documents on transmissible spongiform encephalopathies (TSE) and realised that if Britons could get variant CJD from bovine spongiform encephalopathy (BSE), Americans might get a similar disorder from chronic wasting disease (CWD) the relative of mad cow disease seen among deer and elk in the USA. Although his feverish search did not lead him to the smoking gun linking CWD to a similar disease in North American people, it did uncover a largely disappointing situation.
Singeltary was greatly demoralised at the few attempts to monitor the occurrence of CJD and CWD in the USA. Only a few states have made CJD reportable. Human and animal TSEs should be reportable nationwide and internationally, he complained in a letter to the Journal of the American Medical Association (JAMA 2003; 285: 733). "I hope that the CDC does not continue to expect us to still believe that the 85% plus of all CJD cases which are sporadic are all spontaneous, without route or source."
Until recently, CWD was thought to be confined to the wild in a small region in Colorado. But since early 2002, it has been reported in other areas, including Wisconsin, South Dakota, and the Canadian province of Saskatchewan. Indeed, the occurrence of CWD in states that were not endemic previously increased concern about a widespread outbreak and possible transmission to people and cattle.
To date, experimental studies have proven that the CWD agent can be transmitted to cattle by intracerebral inoculation and that it can cross the mucous membranes of the digestive tract to initiate infection in lymphoid tissue before invasion of the central nervous system. Yet the plausibility of CWD spreading to people has remained elusive.
Part of the problem seems to stem from the US surveillance system. CJD is only reported in those areas known to be endemic foci of CWD. Moreover, US authorities have been criticised for not having performed enough prionic tests in farm deer and elk.
Although in November last year the US Food and Drug Administration issued a directive to state public-health and agriculture officials prohibiting material from CWD-positive animals from being used as an ingredient in feed for any animal species, epidemiological control and research in the USA has been quite different from the situation in the UK and Europe regarding BSE.
"Getting data on TSEs in the USA from the government is like pulling teeth", Singeltary argues. "You get it when they want you to have it, and only what they want you to have."
Norman Foster, director of the Cognitive Disorders Clinic at the University of Michigan (Ann Arbor, MI, USA), says that "current surveillance of prion disease in people in the USA is inadequate to detect whether CWD is occurring in human beings"; adding that, "the cases that we know about are reassuring, because they do not suggest the appearance of a new variant of CJD in the USA or atypical features in patients that might be exposed to CWD. However, until we establish a system that identifies and analyses a high proportion of suspected prion disease cases we will not know for sure". The USA should develop a system modelled on that established in the UK, he points out.
Ali Samii, a neurologist at Seattle VA Medical Center who recently reported the cases of three hunters "two of whom were friends" who died from pathologically confirmed CJD, says that "at present there are insufficient data to claim transmission of CWD into humans"; adding that "[only] by asking [the questions of venison consumption and deer/elk hunting] in every case can we collect suspect cases and look into the plausibility of transmission further". Samii argues that by making both doctors and hunters more aware of the possibility of prions spreading through eating venison, doctors treating hunters with dementia can consider a possible prion disease, and doctors treating CJD patients will know to ask whether they ate venison.
CDC spokesman Ermias Belay says that the CDC "will not be investigating the [Samii] cases because there is no evidence that the men ate CWD-infected meat". He notes that although "the likelihood of CWD jumping the species barrier to infect humans cannot be ruled out 100%" and that "[we] cannot be 100% sure that CWD does not exist in humans& the data seeking evidence of CWD transmission to humans have been very limited".
26 March 2003
Terry S. Singeltary, retired (medically) CJD WATCH
I lost my mother to hvCJD (Heidenhain Variant CJD). I would like to comment on the CDC's attempts to monitor the occurrence of emerging forms of CJD. Asante, Collinge et al [1] have reported that BSE transmission to the 129-methionine genotype can lead to an alternate phenotype that is indistinguishable from type 2 PrPSc, the commonest sporadic CJD. However, CJD and all human TSEs are not reportable nationally. CJD and all human TSEs must be made reportable in every state and internationally. I hope that the CDC does not continue to expect us to still believe that the 85%+ of all CJD cases which are sporadic are all spontaneous, without route/source. We have many TSEs in the USA in both animal and man. CWD in deer/elk is spreading rapidly and CWD does transmit to mink, ferret, cattle, and squirrel monkey by intracerebral inoculation. With the known incubation periods in other TSEs, oral transmission studies of CWD may take much longer. Every victim/family of CJD/TSEs should be asked about route and source of this agent. To prolong this will only spread the agent and needlessly expose others. In light of the findings of Asante and Collinge et al, there should be drastic measures to safeguard the medical and surgical arena from sporadic CJDs and all human TSEs. I only ponder how many sporadic CJDs in the USA are type 2 PrPSc?
***> 2001 FDA CJD TSE Prion Singeltary Submission
Sent: Monday, January 08,2001 3:03 PM
WOW, my submission held up on the www for 17 years, and was proven to be true, and now, it has been removed from the www, the same url does not work anymore and it was just working this year. nothing like the FDA et al cleaning up any evidence of truth with their mad cow debacle and sporadic cjd cover up contineus...so sad$$$
let's review the truth about sporadic cjd shall we;
***> U.S.A. 50 STATE BSE MAD COW CONFERENCE CALL Jan. 9, 2001
[host Richard Barns] and now a question from Terry S. Singeltary of CJD Watch.
[TSS] yes, thank you, U.S. cattle, what kind of guarantee can you give for serum or tissue donor herds?
[no answer, you could hear in the back ground, mumbling and 'we can't. have him ask the question again.]
[host Richard] could you repeat the question?
[TSS] U.S. cattle, what kind of guarantee can you give for serum or tissue donor herds?
[not sure whom ask this] what group are you with?
[TSS] CJD Watch, my Mom died from hvCJD and we are tracking CJD world-wide.
[not sure who is speaking] could you please disconnect Mr. Singeltary
[TSS] you are not going to answer my question?
[not sure whom speaking] NO
[TSS] yes, thank you, U.S. cattle, what kind of guarantee can you give for serum or tissue donor herds?
[no answer, you could hear in the back ground, mumbling and 'we can't. have him ask the question again.]
[host Richard] could you repeat the question?
[TSS] U.S. cattle, what kind of guarantee can you give for serum or tissue donor herds?
[not sure whom ask this] what group are you with?
[TSS] CJD Watch, my Mom died from hvCJD and we are tracking CJD world-wide.
[not sure who is speaking] could you please disconnect Mr. Singeltary
[TSS] you are not going to answer my question?
[not sure whom speaking] NO
2 January 2000 British Medical Journal U.S.
Scientist should be concerned with a CJD epidemic in the U.S., as well
15 November 1999 British Medical Journal hvCJD in the USA * BSE in U.S.
Singeltary on CWD TSE Prion video
Re-Evidence for human transmission of amyloid-β pathology and cerebral amyloid angiopathy
>>> The only tenable public line will be that "more research is required’’ <<<
>>> possibility on a transmissible prion remains open<<<
O.K., so it’s about 23 years later, so somebody please tell me, when is "more research is required’’ enough time for evaluation ?
Re-Evidence for human transmission of amyloid-β pathology and cerebral amyloid angiopathy
Nature 525, 247?250 (10 September 2015) doi:10.1038/nature15369 Received 26 April 2015 Accepted 14 August 2015 Published online 09 September 2015 Updated online 11 September 2015 Erratum (October, 2015)
snip...see full Singeltary Nature comment here;
Alzheimer's disease
let's not forget the elephant in the room. curing Alzheimer's would be a great and wonderful thing, but for starters, why not start with the obvious, lets prove the cause or causes, and then start to stop that. think iatrogenic, friendly fire, or the pass it forward mode of transmission. think medical, surgical, dental, tissue, blood, related transmission. think transmissible spongiform encephalopathy aka tse prion disease aka mad cow type disease...
Commentary: Evidence for human transmission of amyloid-β pathology and cerebral amyloid angiopathy
Self-Propagative Replication of Ab Oligomers Suggests Potential Transmissibility in Alzheimer Disease
*** Singeltary comment PLoS ***
Alzheimer’s disease and Transmissible Spongiform Encephalopathy prion disease, Iatrogenic, what if ?
Posted by flounder on 05 Nov 2014 at 21:27 GMT
IN CONFIDENCE
5 NOVEMBER 1992
TRANSMISSION OF ALZHEIMER TYPE PLAQUES TO PRIMATES
[9. Whilst this matter is not at the moment directly concerned with the iatrogenic CJD cases from hgH, there remains a possibility of litigation here, and this presents an added complication.
There are also results to be made available shortly
(1) concerning a farmer with CJD who had BSE animals,
(2) on the possible transmissibility of Alzheimer’s and
(3) a CMO letter on prevention of iatrogenic CJD transmission in neurosurgery, all of which will serve to increase media interest.]
snip...see full Singeltary Nature comment here;
re-Evidence for human transmission of amyloid-? pathology and cerebral amyloid angiopathy Nature 525, 247?250 (10 September 2015) doi:10.1038/nature15369 Received 26 April 2015 Accepted 14 August 2015 Published online 09 September 2015 Updated online 11 September 2015 Erratum (October, 2015)
I would kindly like to comment on the Nature Paper, the Lancet reply, and the newspaper articles.
First, I applaud Nature, the Scientist and Authors of the Nature paper, for bringing this important finding to the attention of the public domain, and the media for printing said findings.
Secondly, it seems once again, politics is getting in the way possibly of more important Transmissible Spongiform Encephalopathy TSE Prion scientific findings. findings that could have great implications for human health, and great implications for the medical surgical arena. but apparently, the government peer review process, of the peer review science, tries to intervene again to water down said disturbing findings.
where have we all heard this before? it's been well documented via the BSE Inquiry. have they not learned a lesson from the last time?
we have seen this time and time again in England (and other Country's) with the BSE mad cow TSE Prion debacle.
That 'anonymous' Lancet editorial was disgraceful. The editor, Dick Horton is not a scientist.
The pituitary cadavers were very likely elderly and among them some were on their way to CJD or Alzheimer's. Not a bit unusual. Then the recipients, who got pooled extracts injected from thousands of cadavers, were 100% certain to have been injected with both seeds. No surprise that they got both diseases going after thirty year incubations.
That the UK has a "system in place to assist science journalists" to squash embargoed science reports they find 'alarming' is pathetic.
Sounds like the journalists had it right in the first place: 'Alzheimer's may be a transmissible infection' in The Independent to 'You can catch Alzheimer's' in The Daily Mirror or 'Alzheimer's bombshell' in The Daily Express
if not for the journalist, the layperson would not know about these important findings.
where would we be today with sound science, from where we were 30 years ago, if not for the cloak of secrecy and save the industry at all cost mentality?
when you have a peer review system for science, from which a government constantly circumvents, then you have a problem with science, and humans die.
to date, as far as documented body bag count, with all TSE prion named to date, that count is still relatively low (one was too many in my case, Mom hvCJD), however that changes drastically once the TSE Prion link is made with Alzheimer's, the price of poker goes up drastically.
so, who makes that final decision, and how many more decades do we have to wait?
the iatrogenic mode of transmission of TSE prion, the many routes there from, load factor, threshold from said load factor to sub-clinical disease, to clinical disease, to death, much time is there to spread a TSE Prion to anywhere, but whom, by whom, and when, do we make that final decision to do something about it globally? how many documented body bags does it take? how many more decades do we wait? how many names can we make up for one disease, TSE prion?
Professor Collinge et al, and others, have had troubles in the past with the Government meddling in scientific findings, that might in some way involve industry, never mind human and or animal health.
FOR any government to continue to circumvent science for monetary gain, fear factor, or any reason, shame, shame on you.
in my opinion, it's one of the reasons we are at where we are at to date, with regards to the TSE Prion disease science i.e. money, industry, politics, then comes science, in that order.
greed, corporate, lobbyist there from, and government, must be removed from the peer review process of sound science, it's bad enough having them in the pharmaceutical aspect of healthcare policy making, in my opinion.
my mother died from confirmed hvCJD, and her brother (my uncle) Alzheimer's of some type (no autopsy?). just made a promise, never forget, and never let them forget, before I do.
I kindly wish to remind the public of the past, and a possible future we all hopes never happens again. ...
2012
Alzheimer’s disease and Transmissible Spongiform Encephalopathy prion disease, Iatrogenic, what if ?
Background
Alzheimer’s disease and Transmissible Spongiform Encephalopathy disease have both been around a long time, and was discovered in or around the same time frame, early 1900’s. Both diseases are incurable and debilitating brain disease, that are in the end, 100% fatal, with the incubation/clinical period of the Alzheimer’s disease being longer (most of the time) than the TSE prion disease. Symptoms are very similar, and pathology is very similar.
Methods
Through years of research, as a layperson, of peer review journals, transmission studies, and observations of loved ones and friends that have died from both Alzheimer’s and the TSE prion disease i.e. Heidenhain Variant Creutzfelt Jakob Disease CJD.
Results
I propose that Alzheimer’s is a TSE disease of low dose, slow, and long incubation disease, and that Alzheimer’s is Transmissible, and is a threat to the public via the many Iatrogenic routes and sources. It was said long ago that the only thing that disputes this, is Alzheimer’s disease transmissibility, or the lack of. The likelihood of many victims of Alzheimer’s disease from the many different Iatrogenic routes and modes of transmission as with the TSE prion disease.
Conclusions
There should be a Global Congressional Science round table event set up immediately to address these concerns from the many potential routes and sources of the TSE prion disease, including Alzheimer’s disease, and a emergency global doctrine put into effect to help combat the spread of Alzheimer’s disease via the medical, surgical, dental, tissue, and blood arena’s. All human and animal TSE prion disease, including Alzheimer’s should be made reportable in every state, and Internationally, WITH NO age restrictions. Until a proven method of decontamination and autoclaving is proven, and put forth in use universally, in all hospitals and medical, surgical arena’s, or the TSE prion agent will continue to spread. IF we wait until science and corporate politicians wait until politics lets science _prove_ this once and for all, and set forth regulations there from, we will all be exposed to the TSE Prion agents, if that has not happened already.
end...tss
Alzheimer’s disease and Transmissible Spongiform Encephalopathy prion disease, Iatrogenic, what if ?
source references ...end...tss
Hello Nicole,
by all means, please do use my poster. but I thought this was already taken care of, and I could not attend for my poster presentation, therefore, it was not going to be presented. I have some health issues and could not make the trip.
please see old correspondence below...
From: Nicole Sanders Sent: Tuesday, April 10, 2012 5:37 PM To: Terry S. Singeltary Sr. Subject: RE: re-submission
Dear Terry,
The decline of proposal number 30756 is registered in the system.. Thank you for your consideration.
Best Regards,
Nicole
Nicole Sanders
Senior Specialist, Membership & Conference Programming
______________________________________
From: xxxx
To: Terry Singeltary
Sent: Saturday, December 05, 2009 9:09 AM
Subject: 14th ICID - abstract accepted for 'International Scientific Exchange'
Your preliminary abstract number: 670
Dear Mr. Singeltary,
On behalf of the Scientific Committee, I am pleased to inform you that your abstract
'Transmissible Spongiform encephalopathy (TSE) animal and human TSE in North America update October 2009'
WAS accepted for inclusion in the INTERNATIONAL SCIENTIFIC EXCHANGE (ISE) section of the 14th International Congress on Infectious Diseases. Accordingly, your abstract will be included in the "Intl. Scientific Exchange abstract CD-rom" of the Congress which will be distributed to all participants.
Abstracts accepted for INTERNATIONAL SCIENTIFIC EXCHANGE are NOT PRESENTED in the oral OR poster sessions.
Your abstract below was accepted for: INTERNATIONAL SCIENTIFIC EXCHANGE
#0670: Transmissible Spongiform encephalopathy (TSE) animal and human TSE in North America update October 2009
Author: T. Singeltary; Bacliff, TX/US
Topic: Emerging Infectious Diseases Preferred type of presentation: International Scientific Exchange
This abstract has been ACCEPTED.
#0670: Transmissible Spongiform encephalopathy (TSE) animal and human TSE in North America update October 2009
Authors: T. Singeltary; Bacliff, TX/US
Title: Transmissible Spongiform encephalopathy (TSE) animal and human TSE in North America update October 2009
Body: Background
An update on atypical BSE and other TSE in North America. Please remember, the typical U.K. c-BSE, the atypical l-BSE (BASE), and h-BSE have all been documented in North America, along with the typical scrapie's, and atypical Nor-98 Scrapie, and to date, 2 different strains of CWD, and also TME. All these TSE in different species have been rendered and fed to food producing animals for humans and animals in North America (TSE in cats and dogs ?), and that the trading of these TSEs via animals and products via the USA and Canada has been immense over the years, decades.
Methods
12 years independent research of available data
Results
I propose that the current diagnostic criteria for human TSEs only enhances and helps the spreading of human TSE from the continued belief of the UKBSEnvCJD only theory in 2009. With all the science to date refuting it, to continue to validate this old myth, will only spread this TSE agent through a multitude of potential routes and sources i..e. consumption, medical i.e., surgical, blood, dental, endoscopy, optical, nutritional supplements, cosmetics etc.
Conclusion
I would like to submit a review of past CJD surveillance in the USA, and the urgent need to make all human TSE in the USA a reportable disease, in every state, of every age group, and to make this mandatory immediately without further delay. The ramifications of not doing so will only allow this agent to spread further in the medical, dental, surgical arena's. Restricting the reporting of CJD and or any human TSE is NOT scientific. Iatrogenic CJD knows NO age group, TSE knows no boundaries.
I propose as with Aguzzi, Asante, Collinge, Caughey, Deslys, Dormont, Gibbs, Gajdusek, Ironside, Manuelidis, Marsh, et al and many more, that the world of TSE Transmissible Spongiform Encephalopathy is far from an exact science, but there is enough proven science to date that this myth should be put to rest once and for all, and that we move forward with a new classification for human and animal TSE that would properly identify the infected species, the source species, and then the route.
Keywords: Transmissible Spongiform Encephalopathy Creutzfeldt Jakob Disease Prion
page 114 ;
http://ww2.isid.org/Downloads/14th_ICID_ISE_Abstracts.pdf
http://www.isid.org/14th_icid/
http://www.isid.org/publications/ICID_Archive.shtml
http://ww2.isid.org/Downloads/IMED2009_AbstrAuth.pdf
Background
Alzheimer’s disease and Transmissible Spongiform Encephalopathy disease have both been around a long time, and was discovered in or around the same time frame, early 1900’s. Both diseases are incurable and debilitating brain disease, that are in the end, 100% fatal, with the incubation/clinical period of the Alzheimer’s disease being longer (most of the time) than the TSE prion disease. Symptoms are very similar, and pathology is very similar.
Methods
Through years of research, as a layperson, of peer review journals, transmission studies, and observations of loved ones and friends that have died from both Alzheimer’s and the TSE prion disease i.e. Heidenhain Variant Creutzfelt Jakob Disease CJD.
Results
I propose that Alzheimer’s is a TSE disease of low dose, slow, and long incubation disease, and that Alzheimer’s is Transmissible, and is a threat to the public via the many Iatrogenic routes and sources. It was said long ago that the only thing that disputes this, is Alzheimer’s disease transmissibility, or the lack of. The likelihood of many victims of Alzheimer’s disease from the many different Iatrogenic routes and modes of transmission as with the TSE prion disease.
Conclusions
There should be a Global Congressional Science round table event set up immediately to address these concerns from the many potential routes and sources of the TSE prion disease, including Alzheimer’s disease, and a emergency global doctrine put into effect to help combat the spread of Alzheimer’s disease via the medical, surgical, dental, tissue, and blood arena’s. All human and animal TSE prion disease, including Alzheimer’s should be made reportable in every state, and Internationally, WITH NO age restrictions. Until a proven method of decontamination and autoclaving is proven, and put forth in use universally, in all hospitals and medical, surgical arena’s, or the TSE prion agent will continue to spread. IF we wait until science and corporate politicians wait until politics lets science _prove_ this once and for all, and set forth regulations there from, we will all be exposed to the TSE Prion agents, if that has not happened already.
end...tss
Alzheimer’s disease and Transmissible Spongiform Encephalopathy prion disease, Iatrogenic, what if ?
source references ...end...tss
Hello Nicole,
by all means, please do use my poster. but I thought this was already taken care of, and I could not attend for my poster presentation, therefore, it was not going to be presented. I have some health issues and could not make the trip.
please see old correspondence below...
From: Nicole Sanders Sent: Tuesday, April 10, 2012 5:37 PM To: Terry S. Singeltary Sr. Subject: RE: re-submission
Dear Terry,
The decline of proposal number 30756 is registered in the system.. Thank you for your consideration.
Best Regards,
Nicole
Nicole Sanders
Senior Specialist, Membership & Conference Programming
______________________________________
From: xxxx
To: Terry Singeltary
Sent: Saturday, December 05, 2009 9:09 AM
Subject: 14th ICID - abstract accepted for 'International Scientific Exchange'
Your preliminary abstract number: 670
Dear Mr. Singeltary,
On behalf of the Scientific Committee, I am pleased to inform you that your abstract
'Transmissible Spongiform encephalopathy (TSE) animal and human TSE in North America update October 2009'
WAS accepted for inclusion in the INTERNATIONAL SCIENTIFIC EXCHANGE (ISE) section of the 14th International Congress on Infectious Diseases. Accordingly, your abstract will be included in the "Intl. Scientific Exchange abstract CD-rom" of the Congress which will be distributed to all participants.
Abstracts accepted for INTERNATIONAL SCIENTIFIC EXCHANGE are NOT PRESENTED in the oral OR poster sessions.
Your abstract below was accepted for: INTERNATIONAL SCIENTIFIC EXCHANGE
#0670: Transmissible Spongiform encephalopathy (TSE) animal and human TSE in North America update October 2009
Author: T. Singeltary; Bacliff, TX/US
Topic: Emerging Infectious Diseases Preferred type of presentation: International Scientific Exchange
This abstract has been ACCEPTED.
#0670: Transmissible Spongiform encephalopathy (TSE) animal and human TSE in North America update October 2009
Authors: T. Singeltary; Bacliff, TX/US
Title: Transmissible Spongiform encephalopathy (TSE) animal and human TSE in North America update October 2009
Body: Background
An update on atypical BSE and other TSE in North America. Please remember, the typical U.K. c-BSE, the atypical l-BSE (BASE), and h-BSE have all been documented in North America, along with the typical scrapie's, and atypical Nor-98 Scrapie, and to date, 2 different strains of CWD, and also TME. All these TSE in different species have been rendered and fed to food producing animals for humans and animals in North America (TSE in cats and dogs ?), and that the trading of these TSEs via animals and products via the USA and Canada has been immense over the years, decades.
Methods
12 years independent research of available data
Results
I propose that the current diagnostic criteria for human TSEs only enhances and helps the spreading of human TSE from the continued belief of the UKBSEnvCJD only theory in 2009. With all the science to date refuting it, to continue to validate this old myth, will only spread this TSE agent through a multitude of potential routes and sources i..e. consumption, medical i.e., surgical, blood, dental, endoscopy, optical, nutritional supplements, cosmetics etc.
Conclusion
I would like to submit a review of past CJD surveillance in the USA, and the urgent need to make all human TSE in the USA a reportable disease, in every state, of every age group, and to make this mandatory immediately without further delay. The ramifications of not doing so will only allow this agent to spread further in the medical, dental, surgical arena's. Restricting the reporting of CJD and or any human TSE is NOT scientific. Iatrogenic CJD knows NO age group, TSE knows no boundaries.
I propose as with Aguzzi, Asante, Collinge, Caughey, Deslys, Dormont, Gibbs, Gajdusek, Ironside, Manuelidis, Marsh, et al and many more, that the world of TSE Transmissible Spongiform Encephalopathy is far from an exact science, but there is enough proven science to date that this myth should be put to rest once and for all, and that we move forward with a new classification for human and animal TSE that would properly identify the infected species, the source species, and then the route.
Keywords: Transmissible Spongiform Encephalopathy Creutzfeldt Jakob Disease Prion
page 114 ;
http://ww2.isid.org/Downloads/14th_ICID_ISE_Abstracts.pdf
http://www.isid.org/14th_icid/
http://www.isid.org/publications/ICID_Archive.shtml
http://ww2.isid.org/Downloads/IMED2009_AbstrAuth.pdf
P132 Aged cattle brain displays Alzheimer’s-like pathology that can be propagated in a prionlike manner
Ines Moreno-Gonzalez (1), George Edwards III (1), Rodrigo Morales (1), Claudia Duran-Aniotz (1), Mercedes Marquez (2), Marti Pumarola (2), Claudio Soto (1)
(1) Mitchel Center for Alzheimer´s Disease and Related Brain Disorders, Department of Neurology, University of Texas Health Science Center at Houston, Houston, TX, USA (2) Animal Tissue Bank of Catalunya (BTAC), Department of Animal Medicine and Surgery, Veterinary Faculty, Universitat Autónoma de Barcelona, Bellaterra (Cerdanyola del Valles), Barcelona, Spain.
Amyloid beta (Ab) and hyperphosphorylated tau (ptau) are the proteins undergoing misfolding in Alzheimer‘s disease (AD). Recent studies have shown that brain homogenates rich in amyloid aggregates are able to seed the misfolding and aggregation of amyloidogenic proteins inducing an earlier onset of the disease in mouse models of AD. This seeding behavior is analogous to the disease transmission by propagation of prion protein misfolding observed in prion diseases. Prion diseases can be transmitted across species by inoculation of the misfolded prion protein from one specie into an appropriate host. For example, material from cattle affected by bovine spongiform encephalopathy can be propagate in humans inducing variant Creutzfeldt-Jakob disease. In this study, we analyzed the presence of AD-related protein aggregates in the brain of old cows and investigated whether these aggregates are capable to induce pathology in animal models of AD. We observed that many of the typical hallmarks detected in human AD brains, including Ab aggregates and tangles, were present in cow brains. When cattle tissue containing Ab aggregates or ptau were intracerebrally inoculated into APP/PS1 or P301S mice, we observed an acceleration of brain misfolded protein deposition and faster cognitive impairment compared to controls. However, when the material was orally inoculated, no effect was observed. These results may contribute to uncover a previously unsuspected etiology surrounding some cases of sporadic AD. However, the early and controversial stage of the field of prion-like transmission in non-prion diseases added to the artificial nature of the animal models utilized for these studies, indicate that extrapolation of the results to humans should not be done without further experiments.
P75 Determining transmissibility and proteome changes associated with abnormal bovine prionopathy
Dudas S (1,2), Seuberlich T (3), Czub S (1,2)
1. Canadian Food Inspection Agency, NCAD Lethbridge Laboratory, Canada 2. University of Calgary, Canada 3. University of Bern, Switzerland.
In prion diseases, it is believed that altered protein conformation encodes for different pathogenic strains. Currently 3 different strains of bovine spongiform encephalopathy (BSE) are confirmed. Diagnostic tests for BSE are able to identify animals infected with all 3 strains, however, several diagnostic laboratories have reported samples with inconclusive results which are challenging to classify. It was suggested that these may be novel strains of BSE; to determine transmissibility, brain material from index cases were inoculated into cattle.
In the first passage, cattle were intra-cranially challenged with brain homogenate from 2 Swiss animals with abnormal prionopathy. The challenged cattle incubated for 3 years and were euthanized with no clinical signs of neurologic disease. Animals were negative when tested on validated diagnostic tests but several research methods demonstrated changes in the prion conformation in these cattle, including density gradient centrifugation and immunohistochemistry. Currently, samples from the P1 animals are being tested for changes in protein levels using 2-D Fluorescence Difference Gel Electrophoresis (2D DIGE) and mass spectrometry. It is anticipated that, if a prionopathy is present, this approach should identify pathways and targets to decipher the source of altered protein conformation. In addition, a second set of cattle have been challenged with brain material from the first passage. Ideally, these cattle will be given a sufficient incubation period to provide a definitive answer to the question of transmissibility.
=====prion 2018===
P132 Aged cattle brain displays Alzheimer’s-like pathology that can be propagated in a prionlike manner
Ines Moreno-Gonzalez (1), George Edwards III (1), Rodrigo Morales (1), Claudia Duran-Aniotz (1), Mercedes Marquez (2), Marti Pumarola (2), Claudio Soto (1)
(1) Mitchel Center for Alzheimer´s Disease and Related Brain Disorders, Department of Neurology, University of Texas Health Science Center at Houston, Houston, TX, USA (2) Animal Tissue Bank of Catalunya (BTAC), Department of Animal Medicine and Surgery, Veterinary Faculty, Universitat Autónoma de Barcelona, Bellaterra (Cerdanyola del Valles), Barcelona, Spain.
Amyloid beta (Ab) and hyperphosphorylated tau (ptau) are the proteins undergoing misfolding in Alzheimer‘s disease (AD). Recent studies have shown that brain homogenates rich in amyloid aggregates are able to seed the misfolding and aggregation of amyloidogenic proteins inducing an earlier onset of the disease in mouse models of AD. This seeding behavior is analogous to the disease transmission by propagation of prion protein misfolding observed in prion diseases. Prion diseases can be transmitted across species by inoculation of the misfolded prion protein from one specie into an appropriate host. For example, material from cattle affected by bovine spongiform encephalopathy can be propagate in humans inducing variant Creutzfeldt-Jakob disease. In this study, we analyzed the presence of AD-related protein aggregates in the brain of old cows and investigated whether these aggregates are capable to induce pathology in animal models of AD. We observed that many of the typical hallmarks detected in human AD brains, including Ab aggregates and tangles, were present in cow brains. When cattle tissue containing Ab aggregates or ptau were intracerebrally inoculated into APP/PS1 or P301S mice, we observed an acceleration of brain misfolded protein deposition and faster cognitive impairment compared to controls. However, when the material was orally inoculated, no effect was observed. These results may contribute to uncover a previously unsuspected etiology surrounding some cases of sporadic AD. However, the early and controversial stage of the field of prion-like transmission in non-prion diseases added to the artificial nature of the animal models utilized for these studies, indicate that extrapolation of the results to humans should not be done without further experiments.
P75 Determining transmissibility and proteome changes associated with abnormal bovine prionopathy
Dudas S (1,2), Seuberlich T (3), Czub S (1,2)
1. Canadian Food Inspection Agency, NCAD Lethbridge Laboratory, Canada 2. University of Calgary, Canada 3. University of Bern, Switzerland.
In prion diseases, it is believed that altered protein conformation encodes for different pathogenic strains. Currently 3 different strains of bovine spongiform encephalopathy (BSE) are confirmed. Diagnostic tests for BSE are able to identify animals infected with all 3 strains, however, several diagnostic laboratories have reported samples with inconclusive results which are challenging to classify. It was suggested that these may be novel strains of BSE; to determine transmissibility, brain material from index cases were inoculated into cattle.
In the first passage, cattle were intra-cranially challenged with brain homogenate from 2 Swiss animals with abnormal prionopathy. The challenged cattle incubated for 3 years and were euthanized with no clinical signs of neurologic disease. Animals were negative when tested on validated diagnostic tests but several research methods demonstrated changes in the prion conformation in these cattle, including density gradient centrifugation and immunohistochemistry. Currently, samples from the P1 animals are being tested for changes in protein levels using 2-D Fluorescence Difference Gel Electrophoresis (2D DIGE) and mass spectrometry. It is anticipated that, if a prionopathy is present, this approach should identify pathways and targets to decipher the source of altered protein conformation. In addition, a second set of cattle have been challenged with brain material from the first passage. Ideally, these cattle will be given a sufficient incubation period to provide a definitive answer to the question of transmissibility.
=====prion 2018===
END...TSS
IBNC BSE TSE Prion mad cow disease
***however in 1 C-type challenged animal, Prion 2015 Poster Abstracts S67 PrPsc was not detected using rapid tests for BSE.
***Subsequent testing resulted in the detection of pathologic lesion in unusual brain location and PrPsc detection by PMCA only.
*** IBNC Tauopathy or TSE Prion disease, it appears, no one is sure ***
Posted by Terry S. Singeltary Sr. on 03 Jul 2015 at 16:53 GMT
THURSDAY, SEPTEMBER 27, 2018
Amydis Awarded Prion Disease Grant from NIH
THURSDAY, OCTOBER 04, 2018
***> Cervid to human prion transmission 5R01NS088604-04 Update
Extra View
Sunday, November 23, 2014
Confirmed Variant Creutzfeldt-Jakob Disease (variant CJD) Case in Texas
Updated: October 7, 2014
CDC and the Texas Department of State Health Services (DSHS) have completed the investigation of the recently reported fourth vCJD case in the United States. It confirmed that the case was in a US citizen born outside the Americas and indicated that the patient's exposure to the BSE/vCJD agent most likely occurred before he moved to the United States; the patient had resided in Kuwait, Russia and Lebanon. The completed investigation did not support the patient's having had extended travel to European countries, including the United Kingdom, or travel to Saudi Arabia. The specific overseas country where this patient’s infection occurred is less clear largely because the investigation did not definitely link him to a country where other known vCJD cases likely had been infected.
https://www.cdc.gov/ncidod/dvrd/vcjd/other/confirmed-case-in-texas.htm
https://vcjd.blogspot.com/2014/11/confirmed-variant-creutzfeldt-jakob.html
4. The link between BSE and vCJD
USA CJD TSE Tables of Cases Examined National Prion Disease Pathology Surveillance Center Cases Examined May 1, 2018
http://prionunitusaupdate.blogspot.com/2018/07/usa-cjd-tse-tables-of-cases-examined.html
Unexpected prion phenotypes in experimentally transfused animals: predictive models for humans?
Emmanuel E. Comoy, Jacqueline Mikol & Jean-Philippe Deslys
Pages 162-169 | Received 06 Jul 2018, Accepted 24 Jul 2018, Accepted author version posted online: 06 Aug 2018, Published online: 16 Aug 2018
ABSTRACT
The recently reevaluated high prevalence of healthy carriers (1/2,000 in UK) of variant Creutzfeldt-Jakob Disease (v-CJD), whose blood might be infectious, suggests that the evolution of this prion disease might not be under full control as expected.
After experimental transfusion of macaques and conventional mice with blood derived from v-CJD exposed (human and animal) individuals, we confirmed in these both models the transmissibility of v-CJD, but we also observed unexpected neurological syndromes transmissible by transfusion: despite their prion etiology confirmed through transmission experiments, these original cases would escape classical prion diagnosis, notably in the absence of detectable abnormal PrP with current techniques.
It is noteworthy that macaques developed an original, yet undescribed myelopathic syndrome associating demyelination and pseudo-necrotic lesions of spinal cord, brainstem and optical tract without affecting encephalon, which is rather evocative of spinal cord disease than prion disease in human medicine.
These observations strongly suggest that the spectrum of human prion diseases may extend the current field restricted to the phenotypes associated to protease-resistant PrP, and may notably include spinal cord diseases.
KEYWORDS: (5-10): prion, vCJD, macaque, mouse, spinal cord disease, myelopathy, transfusion, abnormal PrP, demyelination, spongiform change
This article refers to:
Additional information
Funding
The original laboratory work was funded by European Commission [Fifth Framework Programme, QLK1-CT-2002-01096], French Research Funding Agency (Agence Nationale de la Recherche, ANR) [ANR-10-BLAN-1330 01], Health Canada [4500216567] and MacoPharma.
TUESDAY, AUGUST 7, 2018
Unexpected prion phenotypes in experimentally transfused animals: predictive models for humans?
MONDAY, NOVEMBER 06, 2017
Experimental transfusion of variant CJD-infected blood reveals previously uncharacterised prion disorder in mice and macaque
HUMAN MAD COW DISEASE nvCJD TEXAS CASE NOT LINKED TO EUROPEAN TRAVEL CDC
Sunday, November 23, 2014
Confirmed Variant Creutzfeldt-Jakob Disease (variant CJD) Case in Texas
Updated: October 7, 2014
CDC and the Texas Department of State Health Services (DSHS) have completed the investigation of the recently reported fourth vCJD case in the United States. It confirmed that the case was in a US citizen born outside the Americas and indicated that the patient's exposure to the BSE/vCJD agent most likely occurred before he moved to the United States; the patient had resided in Kuwait, Russia and Lebanon. The completed investigation did not support the patient's having had extended travel to European countries, including the United Kingdom, or travel to Saudi Arabia. The specific overseas country where this patient’s infection occurred is less clear largely because the investigation did not definitely link him to a country where other known vCJD cases likely had been infected.
https://www.cdc.gov/ncidod/dvrd/vcjd/other/confirmed-case-in-texas.htm
https://vcjd.blogspot.com/2014/11/confirmed-variant-creutzfeldt-jakob.html
WEDNESDAY, SEPTEMBER 26, 2018
A new variant of Creutzfeldt-Jakob disease in the UK 1995 revisited 2018 a review of science
THURSDAY, OCTOBER 04, 2018
National Prion Disease Pathology Surveillance Center Cases Examined¹ (September 18, 2018)
FRIDAY, OCTOBER 05, 2018
More Politicians and Very Young People Struck Down With Creutzfeldt Jakob Disease CJD mad cow type TSE Prion USA
THURSDAY, SEPTEMBER 27, 2018
Amydis Awarded Prion Disease Grant from NIH
Volume 2: Science
4. The link between BSE and vCJD
4.29 The evidence discussed above that vCJD is caused by BSE seems overwhelming. Uncertainties exist about the cause of CJD in farmers, their wives and in several abattoir workers. It seems that farmers at least might be at higher risk than others in the general population. 1 Increased ascertainment (ie, increased identification of cases as a result of greater awareness of the condition) seems unlikely, as other groups exposed to risk, such as butchers and veterinarians, do not appear to have been affected.
***The CJD in farmers seems to be similar to other sporadic CJD in age of onset, in respect to glycosylation patterns, and in strain-typing in experimental mice.
***Some farmers are heterozygous for the methionine/valine variant at codon 129, and their lymphoreticular system (LRS) does not contain the high levels of PrPSc found in vCJD.
*** It remains a remote possibility that when older people contract CJD from BSE the resulting phenotype is like sporadic CJD and is distinct from the vCJD phenotype in younger people.
BSEINQUIRY
***>Some farmers are heterozygous for the methionine/valine variant at codon 129, and their lymphoreticular system (LRS) does not contain the high levels of PrPSc found in vCJD.<***
SATURDAY, JUNE 23, 2018
Diagnosis of Methionine/Valine Variant Creutzfeldt-Jakob Disease by Protein Misfolding Cyclic Amplification
Volume 24, Number 7—July 2018
2006-2007
HUMAN and ANIMAL TSE Classifications i.e. mad cow disease and the UKBSEnvCJD only theory
TSEs have been rampant in the USA for decades in many species, and they all have been rendered and fed back to animals for human/animal consumption. I propose that the current diagnostic criteria for human TSEs only enhances and helps the spreading of human TSE from the continued belief of the UKBSEnvCJD only theory in 2007. With all the science to date refuting it, to continue to validate this myth, will only spread this TSE agent through a multitude of potential routes and sources i.e. consumption, surgical, blood, medical, cosmetics etc. I propose as with Aguzzi, Asante, Collinge, Caughey, Deslys, Dormont, Gibbs, Ironside, Manuelidis, Marsh, et al and many more, that the world of TSE Transmissible Spongiform Encephalopathy is far from an exact science, but there is enough proven science to date that this myth should be put to rest once and for all, and that we move forward with a new classification for human and animal TSE that would properly identify the infected species, the source species, and then the route. This would further have to be broken down to strain of species and then the route of transmission would further have to be broken down. Accumulation and Transmission are key to the threshold from subclinical to clinical disease, and of that, I even believe that physical and or blunt trauma may play a role of onset of clinical symptoms in some cases, but key to all this, is to stop the amplification and transmission of this agent, the spreading of, no matter what strain. BUT, to continue with this myth that the U.K. strain of BSE one strain in cows, and the nv/v CJD, one strain in humans, and that all the rest of human TSE is one single strain i.e. sporadic CJD (when to date there are 6 different phenotypes of sCJD), and that no other animal TSE transmits to humans, to continue with this masquerade will only continue to spread, expose, and kill, who knows how many more in the years and decades to come. ONE was enough for me, My Mom, hvCJD, DOD 12/14/97 confirmed, which is nothing more than another mans name added to CJD, like CJD itself, Jakob and Creutzfeldt, or Gerstmann-Straussler-Scheinker syndrome, just another CJD or human TSE, named after another human. WE are only kidding ourselves with the current diagnostic criteria for human and animal TSE, especially differentiating between the nvCJD vs the sporadic CJD strains and then the GSS strains and also the FFI fatal familial insomnia strains or the ones that mimics one or the other of those TSE? Tissue infectivity and strain typing of the many variants of the human and animal TSEs are paramount in all variants of all TSE. There must be a proper classification that will differentiate between all these human TSE in order to do this. With the CDI and other more sensitive testing coming about, I only hope that my proposal will some day be taken seriously.
My name is Terry S. Singeltary Sr. and I am no scientist, no doctor and have no PhDs, but have been independently researching human and animal TSEs since the death of my Mother to the Heidenhain Variant of Creutzfeldt Jakob Disease on December 14, 1997 'confirmed'. ...END
Diagnosis and Reporting of Creutzfeldt-Jakob Disease
Singeltary, Sr et al. JAMA.2001; 285: 733-734. Vol. 285 No. 6, February 14, 2001 JAMA Diagnosis and Reporting of Creutzfeldt-Jakob Disease
To the Editor:
In their Research Letter, Dr Gibbons and colleagues1 reported that the annual US death rate due to Creutzfeldt-Jakob disease (CJD) has been stable since 1985. These estimates, however, are based only on reported cases, and do not include misdiagnosed or preclinical cases. It seems to me that misdiagnosis alone would drastically change these figures. An unknown number of persons with a diagnosis of Alzheimer disease in fact may have CJD, although only a small number of these patients receive the postmortem examination necessary to make this diagnosis. Furthermore, only a few states have made CJD reportable. Human and animal transmissible spongiform encephalopathies should be reportable nationwide and internationally.
Terry S. Singeltary, Sr Bacliff, Tex
1. Gibbons RV, Holman RC, Belay ED, Schonberger LB. Creutzfeldt-Jakob disease in the United States: 1979-1998. JAMA. 2000;284:2322-2323.
Extra View
Unexpected prion phenotypes in experimentally transfused animals: predictive models for humans?
Emmanuel E. Comoy, Jacqueline Mikol & Jean-Philippe Deslys
Pages 162-169 | Received 06 Jul 2018, Accepted 24 Jul 2018, Accepted author version posted online: 06 Aug 2018, Published online: 16 Aug 2018
ABSTRACT
The recently reevaluated high prevalence of healthy carriers (1/2,000 in UK) of variant Creutzfeldt-Jakob Disease (v-CJD), whose blood might be infectious, suggests that the evolution of this prion disease might not be under full control as expected.
After experimental transfusion of macaques and conventional mice with blood derived from v-CJD exposed (human and animal) individuals, we confirmed in these both models the transmissibility of v-CJD, but we also observed unexpected neurological syndromes transmissible by transfusion: despite their prion etiology confirmed through transmission experiments, these original cases would escape classical prion diagnosis, notably in the absence of detectable abnormal PrP with current techniques.
It is noteworthy that macaques developed an original, yet undescribed myelopathic syndrome associating demyelination and pseudo-necrotic lesions of spinal cord, brainstem and optical tract without affecting encephalon, which is rather evocative of spinal cord disease than prion disease in human medicine.
These observations strongly suggest that the spectrum of human prion diseases may extend the current field restricted to the phenotypes associated to protease-resistant PrP, and may notably include spinal cord diseases.
KEYWORDS: (5-10): prion, vCJD, macaque, mouse, spinal cord disease, myelopathy, transfusion, abnormal PrP, demyelination, spongiform change
This article refers to:
Additional information
Funding
The original laboratory work was funded by European Commission [Fifth Framework Programme, QLK1-CT-2002-01096], French Research Funding Agency (Agence Nationale de la Recherche, ANR) [ANR-10-BLAN-1330 01], Health Canada [4500216567] and MacoPharma.
TUESDAY, AUGUST 7, 2018
Unexpected prion phenotypes in experimentally transfused animals: predictive models for humans?
MONDAY, NOVEMBER 06, 2017
Experimental transfusion of variant CJD-infected blood reveals previously uncharacterised prion disorder in mice and macaque
Volume 2: Science
4. The link between BSE and vCJD4.29 The evidence discussed above that vCJD is caused by BSE seems overwhelming.. Uncertainties exist about the cause of CJD in farmers, their wives and in several abattoir workers. It seems that farmers at least might be at higher risk than others in the general population. 1 Increased ascertainment (ie, increased identification of cases as a result of greater awareness of the condition) seems unlikely, as other groups exposed to risk, such as butchers and veterinarians, do not appear to have been affected.***The CJD in farmers seems to be similar to other sporadic CJD in age of onset, in respect to glycosylation patterns, and in strain-typing in experimental mice.***Some farmers are heterozygous for the methionine/valine variant at codon 129, and their lymphoreticular system (LRS) does not contain the high levels of PrPSc found in vCJD.*** It remains a remote possibility that when older people contract CJD from BSE the resulting phenotype is like sporadic CJD and is distinct from the vCJD phenotype in younger people.BSEINQUIRY***>Some farmers are heterozygous for the methionine/valine variant at codon 129, and their lymphoreticular system (LRS) does not contain the high levels of PrPSc found in vCJD.<***SATURDAY, JUNE 23, 2018***> Diagnosis of Methionine/Valine Variant Creutzfeldt-Jakob Disease by Protein Misfolding Cyclic AmplificationVolume 24, Number 7—July 20182006-2007HUMAN and ANIMAL TSE Classifications i.e. mad cow disease and the UKBSEnvCJD only theoryTSEs have been rampant in the USA for decades in many species, and they all have been rendered and fed back to animals for human/animal consumption. I propose that the current diagnostic criteria for human TSEs only enhances and helps the spreading of human TSE from the continued belief of the UKBSEnvCJD only theory in 2007. With all the science to date refuting it, to continue to validate this myth, will only spread this TSE agent through a multitude of potential routes and sources i.e. consumption, surgical, blood, medical, cosmetics etc. I propose as with Aguzzi, Asante, Collinge, Caughey, Deslys, Dormont, Gibbs, Ironside, Manuelidis, Marsh, et al and many more, that the world of TSE Transmissible Spongiform Encephalopathy is far from an exact science, but there is enough proven science to date that this myth should be put to rest once and for all, and that we move forward with a new classification for human and animal TSE that would properly identify the infected species, the source species, and then the route. This would further have to be broken down to strain of species and then the route of transmission would further have to be broken down. Accumulation and Transmission are key to the threshold from subclinical to clinical disease, and of that, I even believe that physical and or blunt trauma may play a role of onset of clinical symptoms in some cases, but key to all this, is to stop the amplification and transmission of this agent, the spreading of, no matter what strain. BUT, to continue with this myth that the U.K. strain of BSE one strain in cows, and the nv/v CJD, one strain in humans, and that all the rest of human TSE is one single strain i.e. sporadic CJD (when to date there are 6 different phenotypes of sCJD), and that no other animal TSE transmits to humans, to continue with this masquerade will only continue to spread, expose, and kill, who knows how many more in the years and decades to come. ONE was enough for me, My Mom, hvCJD, DOD 12/14/97 confirmed, which is nothing more than another mans name added to CJD, like CJD itself, Jakob and Creutzfeldt, or Gerstmann-Straussler-Scheinker syndrome, just another CJD or human TSE, named after another human. WE are only kidding ourselves with the current diagnostic criteria for human and animal TSE, especially differentiating between the nvCJD vs the sporadic CJD strains and then the GSS strains and also the FFI fatal familial insomnia strains or the ones that mimics one or the other of those TSE? Tissue infectivity and strain typing of the many variants of the human and animal TSEs are paramount in all variants of all TSE. There must be a proper classification that will differentiate between all these human TSE in order to do this. With the CDI and other more sensitive testing coming about, I only hope that my proposal will some day be taken seriously.My name is Terry S. Singeltary Sr. and I am no scientist, no doctor and have no PhDs, but have been independently researching human and animal TSEs since the death of my Mother to the Heidenhain Variant of Creutzfeldt Jakob Disease on December 14, 1997 'confirmed'. ...ENDDiagnosis and Reporting of Creutzfeldt-Jakob DiseaseSingeltary, Sr et al. JAMA.2001; 285: 733-734. Vol. 285 No. 6, February 14, 2001 JAMA Diagnosis and Reporting of Creutzfeldt-Jakob DiseaseTo the Editor:In their Research Letter, Dr Gibbons and colleagues1 reported that the annual US death rate due to Creutzfeldt-Jakob disease (CJD) has been stable since 1985. These estimates, however, are based only on reported cases, and do not include misdiagnosed or preclinical cases. It seems to me that misdiagnosis alone would drastically change these figures. An unknown number of persons with a diagnosis of Alzheimer disease in fact may have CJD, although only a small number of these patients receive the postmortem examination necessary to make this diagnosis. Furthermore, only a few states have made CJD reportable. Human and animal transmissible spongiform encephalopathies should be reportable nationwide and internationally.Terry S. Singeltary, Sr Bacliff, Tex1. Gibbons RV, Holman RC, Belay ED, Schonberger LB. Creutzfeldt-Jakob disease in the United States: 1979-1998. JAMA. 2000;284:2322-2323.Diagnosis and Reporting of Creutzfeldt-Jakob DiseaseSingeltary, Sr et al. JAMA.2001; 285: 733-734.BRITISH MEDICAL JOURNALBMJU.S. Scientist should be concerned with a CJD epidemic in the U.S., as well...02 January 2000Terry S Singeltaryretired
US scientists develop a possible test for BSEBMJ 1999; 319 doi: https://doi.org/10.1136/bmj.319.7220.1312b (Published 13 November 1999) Cite this as: BMJ 1999;319:1312Rapid responses ResponseRe: vCJD in the USA * BSE in U.S.15 November 1999Terry S SingeltaryNAmedically retiredJanuary 28, 2003; 60 (2) VIEWS & REVIEWSMonitoring the occurrence of emerging forms of Creutzfeldt-Jakob disease in the United StatesErmias D. Belay, Ryan A. Maddox, Pierluigi Gambetti, Lawrence B. SchonbergerFirst published January 28, 2003, DOI: https://doi.org/10.1212/01.WNL.0000036913.87823.D6AbstractTransmissible spongiform encephalopathies (TSEs) attracted increased attention in the mid-1980s because of the emergence among UK cattle of bovine spongiform encephalopathy (BSE), which has been shown to be transmitted to humans, causing a variant form of Creutzfeldt-Jakob disease (vCJD). The BSE outbreak has been reported in 19 European countries, Israel, and Japan, and human cases have so far been identified in four European countries, and more recently in a Canadian resident and a US resident who each lived in Britain during the BSE outbreak. To monitor the occurrence of emerging forms of CJD, such as vCJD, in the United States, the Centers for Disease Control and Prevention has been conducting surveillance for human TSEs through several mechanisms, including the establishment of the National Prion Disease Pathology Surveillance Center. Physicians are encouraged to maintain a high index of suspicion for vCJD and use the free services of the pathology center to assess the neuropathology of clinically diagnosed and suspected cases of CJD or other TSEs.Received May 7, 2002. Accepted August 28, 2002.
RE-Monitoring the occurrence of emerging forms of Creutzfeldt-Jakob disease in the United StatesTerry S. Singeltary, retired (medically)Published March 26, 200326 March 2003Terry S. Singeltary, retired (medically) CJD WATCHI lost my mother to hvCJD (Heidenhain Variant CJD). I would like to comment on the CDC's attempts to monitor the occurrence of emerging forms of CJD. Asante, Collinge et al [1] have reported that BSE transmission to the 129-methionine genotype can lead to an alternate phenotype that is indistinguishable from type 2 PrPSc, the commonest sporadic CJD. However, CJD and all human TSEs are not reportable nationally. CJD and all human TSEs must be made reportable in every state and internationally. I hope that the CDC does not continue to expect us to still believe that the 85%+ of all CJD cases which are sporadic are all spontaneous, without route/source. We have many TSEs in the USA in both animal and man. CWD in deer/elk is spreading rapidly and CWD does transmit to mink, ferret, cattle, and squirrel monkey by intracerebral inoculation. With the known incubation periods in other TSEs, oral transmission studies of CWD may take much longer. Every victim/family of CJD/TSEs should be asked about route and source of this agent. To prolong this will only spread the agent and needlessly expose others. In light of the findings of Asante and Collinge et al, there should be drastic measures to safeguard the medical and surgical arena from sporadic CJDs and all human TSEs. I only ponder how many sporadic CJDs in the USA are type 2 PrPSc?Reply to Singletary Ryan A. Maddox, MPH Other Contributors: Published March 26, 2003Mr. Singletary raises several issues related to current Creutzfeldt- Jakob disease (CJD) surveillance activities. Although CJD is not a notifiable disease in most states, its unique characteristics, particularly its invariably fatal outcome within usually a year of onset, make routine mortality surveillance a useful surrogate for ongoing CJD surveillance.[1] In addition, because CJD is least accurately diagnosed early in the course of illness, notifiable-disease surveillance could be less accurate than, if not duplicative of, current mortality surveillance.[1] However, in states where making CJD officially notifiable would meaningfully facilitate the collection of data to monitor for variant CJD (vCJD) or other emerging prion diseases, CDC encourages the designation of CJD as a notifiable disease.[1] Moreover, CDC encourages physicians to report any diagnosed or suspected CJD cases that may be of special public health importance (e.g., vCJD, iatrogenic CJD, unusual CJD clusters).As noted in our article, strong evidence is lacking for a causal link between chronic wasting disease (CWD) of deer and elk and human disease,[2] but only limited data seeking such evidence exist. Overall, the previously published case-control studies that have evaluated environmental sources of infection for sporadic CJD have not consistently identified strong evidence for a common risk factor.[3] However, the power of a case-control study to detect a rare cause of CJD is limited, particularly given the relatively small number of subjects generally involved and its long incubation period, which may last for decades. Because only a very small proportion of the US population has been exposed to CWD, a targeted surveillance and investigation of unusual cases or case clusters of prion diseases among persons at increased risk of exposure to CWD is a more efficient approach to detecting the possible transmission of CWD to humans. In collaboration with appropriate local and state health departments and the National Prion Disease Pathology Surveillance Center, CDC is facilitating or conducting such surveillance and case- investigations, including related laboratory studies to characterize CJD and CWD prions.Mr. Singletary also expresses concern over a recent publication by Asante and colleagues indicating the possibility that some sporadic CJD cases may be attributable to bovine spongiform encephalopathy (BSE).[4] The authors reported that transgenic mice expressing human prion protein homozygous for methionine at codon 129, when inoculated with BSE prions, developed a molecular phenotype consistent with a subtype of sporadic CJD. Although the authors implied that BSE might cause a sporadic CJD-like illness among persons homozygous for methionine, the results of their research with mice do not necessarily directly apply to the transmission of BSE to humans. If BSE causes a sporadic CJD-like illness in humans, an increase in sporadic CJD cases would be expected to first occur in the United Kingdom, where the vast majority of vCJD cases have been reported. In the United Kingdom during 1997 through 2002, however, the overall average annual mortality rate for sporadic CJD was not elevated; it was about 1 case per million population per year. In addition, during this most recent 6-year period following the first published description of vCJD in 1996, there was no increasing trend in the reported annual number of UK sporadic CJD deaths.[3, 5] Furthermore, surveillance in the UK has shown no increase in the proportion of sporadic CJD cases that are homozygous for methionine (Will RG, National CJD Surveillance Unit, United Kingdom, 2003; personal communication).References1. Gibbons RV, Holman RC, Belay ED, Schonberger LB. Diagnosis and reporting of Creutzfeldt-Jakob disease. JAMA 2001;285:733-734.2. Belay ED, Maddox RA, Gambetti P, Schonberger LB. Monitoring the occurrence of emerging forms of Creutzfeldt-Jakob disease in the United States. Neurology 2003;60:176-181.3. Belay ED. Transmissible spongiform encephalopathies in humans. Annu Rev Microbiol 1999;53:283-314.4. Asante EA, Linehan JM, Desbruslais M, et al. BSE prions propagate as either variant CJD-like or sporadic CJD-like prion strains in transgenic mice expressing human prion protein. EMBO J 2002;21:6358-6366.5. The UK Creutzfeldt-Jakob Disease Surveillance Unit. CJD statistics. Available at: http://www.cjd.ed.ac.uk/figures.htm. Accessed February 18, 2003.Competing Interests: None declared.doi:10.1016/S1473-3099(03)00715-1 Copyright © 2003 Published by Elsevier Ltd. NewsdeskTracking spongiform encephalopathies in North AmericaXavier BoschAvailable online 29 July 2003.Volume 3, Issue 8, August 2003, Page 463“My name is Terry S Singeltary Sr, and I live in Bacliff, Texas. I lost my mom to hvCJD (Heidenhain variant CJD) and have been searching for answers ever since. What I have found is that we have not been told the truth. CWD in deer and elk is a small portion of a much bigger problem.” ............................see moms autopsy;TUESDAY, SEPTEMBER 4, 2018USA CJD, BSE, SCRAPIE, CWD, TSE PRION END OF YEAR REPORTS September 4, 2018prepare for the storm...WEDNESDAY, OCTOBER 17, 2018PRICE OF TSE PRION POKER GOES UP spectrum of human prion diseases may extend the current field and may notably include spinal cord diseases
TUESDAY, JULY 31, 2018
USA CJD TSE Tables of Cases Examined National Prion Disease Pathology Surveillance Center Cases Examined May 1, 2018
http://prionunitusaupdate.blogspot.com/2018/07/usa-cjd-tse-tables-of-cases-examined.html
Tuesday, March 20, 2018
Variably protease-sensitive prionopathy (VPSPr), sporadic creutzfeldt jakob disease sCJD, the same disease, what if?
O3 Experimental studies on prion transmission barrier and TSE pathogenesis in large animals
Rosa Bolea(1), Acín C(1)Marín B(1), Hedman C(1), Raksa H(1), Barrio T(1), Otero A(1), LópezPérez O(1), Monleón E(1),Martín-Burriel(1), Monzón M(1), Garza MC(1), Filali H(1),Pitarch JL(1), Garcés M(1), Betancor M(1), GuijarroIM(1), GarcíaM(1), Moreno B(1),Vargas A(1), Vidal E(2), Pumarola M(2), Castilla J(3), Andréoletti O(4), Espinosa JC(5), Torres JM(5), Badiola JJ(1).
1Centro de Investigación en Encefalopatías y Enfermedades Transmisibles Emergentes, VeterinaryFaculty, Universidad de Zaragoza; Zaragoza,Spain.2 RTA, Centre de Recerca en Sanitat Animal (CReSA, IRTA-UAB) 3 4 INRA, ÉcoleVétérinaire, Toulouse, France.5CIC bioGUNE, Prion researchlab, Derio, Spain CISA- INIA, Valdeolmos, Madrid 28130, Spain.
Experimental transmission of Transmissible Spongiform Encephalopathies (TSE) has been understood and related with several factors that could modify the natural development of these diseases. In fact, the behaviour of the natural disease does not match exactly in each animal, being modified by parameters such as the age at infection, the genotype, the breed or the causative strain. Moreover, different TSE strains can target different animal species or tissues, what complicate the prediction of its transmissibility when is tested in a different species of the origin source. The aim of the experimental studies in large animals is to homogenize all those factors, trying to minimize as much as possible variations between individuals. These effects can be flattened by experimental transmission in mice, in which a specific strain can be selected after several passages. With this objective, several experimental studies in large animals have been developed by the presenter research team.
Classical scrapie agent has been inoculated in cow, with the aim of demonstrate the resistance or susceptibility of this species to the first well known TSE; Atypical scrapie has been inoculated in sheep (using several routes of infection), cow and pig, with the objective of evaluating the potential pathogenicity of this strain; Classical Bovine Spongiform Encephalopathy (BSE) has been inoculated in goats aiming to demonstrate if the genetic background of this species could protect against this strain; goat BSE and sheep BSE have been inoculated in goats and pigs respectively to evaluate the effect of species barrier; and finally atypical BSE has been inoculated in cattle to assess the transmissibility properties of this newly introduced strain.
Once the experiments have been carried out on large animal species, a collection of samples from animals studied were inoculated in different types of tg mice overexpressing PrPcin order to study the infectivity of the tissues, and also were studied using PMCA.
In summary, the parameters that have been controlled are the species, the strain, the route of inoculation, the time at infection, the genotype, the age, and the environmental conditions.
To date,
***> eleven of the atypical scrapie intracerebrally inoculated sheep have succumbed to atypical scrapie disease;
***> six pigs to sheep BSE;
***> one cow to classical scrapie;
***> nine goats to goat BSE and
***> five goats to classical BSE.
***> PrPSC has been demonstrated in all cases by immunohistochemistry and western blot.
=====> PRION CONFERENCE 2018
Terry S. Singeltary Sr.
FRIDAY, OCTOBER 19, 2018
Mississippi Chronic Wasting Disease Suspected in a Second White-tailed Deer 10/19/2018 4:03:08 PM From MDWFP
posted by Terry S. Singeltary Sr. at
4:20 PM
![]()
![]()


0 Comments:
Post a Comment
Subscribe to Post Comments [Atom]
<< Home